The definition and classification of cardiomyopathy have evolved considerably in recent years. Cardiomyopathy can be separated into primary (genetic, mixed, or acquired) and secondary categories, which result in varied phenotypes including dilated, hypertrophic, and restrictive patterns. Hypertrophic cardiomyopathy is the most common primary cardiomyopathy and can cause exertional dyspnea, presyncope, atypical chest pain, heart failure, and sudden cardiac death. Dilated cardiomyopathy can be genetic or acquired and typically presents with classic symptoms of heart failure with reduced ejection fraction. Restrictive cardiomyopathy is much less common and often associated with systemic disease. Family physicians should be alert for acquired variants of cardiomyopathy, including peripartum and stress-induced cardiomyopathy, as well as rare variants, such as arrhythmogenic right ventricular dysplasia and left ventricular noncompaction. In addition to history and physical examination, diagnosis of cardiomyopathy includes electrocardiography and echocardiography testing. Treatment may include appropriately staged therapy for heart failure, appropriate activity restriction, evaluation for implantable cardioverter-defibrillator placement, and consideration of heart transplantation in refractory cases. Genetic testing of families is an emerging modality with some potential to augment traditional screening performed by family physicians.
Manifestations of cardiomyopathy range from microscopic alterations in cardiac myocytes to fulminant heart failure with inadequate tissue perfusion, fluid accumulation, and cardiac rhythm dysfunction. Historically, cardiomyopathy, which literally means heart muscle disease, was separated into hypertrophic, dilated, and restrictive categories. However, advances in genomics have made it clear that there is variety in phenotypic expression.1
WHAT IS NEW ON THIS TOPIC: CARDIOMYOPATHY
Pathologies with a known cardiovascular cause, including hypertension, valvular disease, congenital heart disease, and coronary ischemia, are now excluded from the term cardiomyopathy.
An uncommon and recently identified congenital cardiomyopathy is left ventricular noncompaction, a condition of embryonic origin that interferes with the development of mature heart muscle. The disease is defined by significant trabeculation of the myocardium, in addition to development of intertrabecular recesses in the left ventricle.
SORT: KEY RECOMMENDATIONS FOR PRACTICE

| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Heart failure with reduced ejection fraction should be managed according to the most recent American College of Cardiology/American Heart Association guidelines. | C | 5, 6 |
| Hypertrophic cardiomyopathy should be managed according to the most recent American College of Cardiology Foundation/American Heart Association guidelines. | C | 12 |
| An implanted cardioverter-defibrillator should be placed in patients who are at risk of sudden cardiac death. | C | 1 |
| Heart transplantation should be considered if cardiomyopathy is refractory to medical therapy. | C | 8 |
| Patients with cardiomyopathy should be referred for genetic counseling. | C | 44 |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.
The American Heart Association now endorses a classification system that categorizes cardiomyopathy as primary or secondary. In primary cases, the disease process is chiefly confined to the heart. Secondary cardiomyopathy describes conditions in which cardiac involvement occurs as part of a systemic condition. This classification system is imperfect, and there is often overlap.1
Primary cardiomyopathies can be genetic, acquired, or mixed in etiology (Table 1).1 Genetic cardiomyopathies are caused by chromosomal abnormalities that affect the heart. Acquired, not to be confused with secondary, cardiomyopathies involve non-genetic causes that lead to chiefly, or even exclusively, to cardiac complications. In mixed types, a common phenotype is realized through genetic and nongenetic means.1
Table 1. Classification of Primary Cardiomyopathies

| Acquired |
| Myocarditis |
| Peripartum |
| Tachycardia induced |
| Takotsubo (stress induced) |
| Genetic |
| Arrhythmogenic right ventricular dysplasia |
| Hypertrophic |
| Ion channel disorders |
| Left ventricular compaction |
| Mitochondrial myopathies |
| Mixed |
| Dilated |
| Restrictive |
Information from reference 1.
It has commonly been understood that hypertrophic and dilated patterns stem from hypertension and coronary artery disease, respectively. However, pathologies with a specific known cardiovascular cause, including hypertension, valvular disease, congenital heart disease, and coronary ischemia, are now excluded from the term cardiomyopathy. As a result, the term ischemic cardiomyopathy is inaccurate under current nomenclature.1,2 This term is still commonly used in clinical practice, however, and classification continues to evolve.
General Overview
Although they involve a variety of phenotypes and etiologies, the most common cardiomyopathies often present to primary care physicians with similar symptoms. Hypertrophic, dilated, and restrictive cardiomyopathy may each present with signs and symptoms that are common in heart failure with reduced ejection fraction, including peripheral edema, fatigue, orthopnea, dyspnea on exertion, paroxysmal nocturnal dyspnea, presyncope, syncope, and cardiac ischemia.1,3,4 In certain instances, symptoms suggest one type of cardiomyopathy over another. It is important that primary care physicians recognize these symptoms and pursue appropriate diagnostic measures, beginning with electrocardiography and echocardiography. Treatment of symptomatic heart failure should follow current American College of Cardiology/American Heart Association guidelines5,6 (Figure 15–7 ). Pharmacologic therapy may include use of a beta blocker, angiotensin-converting enzyme (ACE) inhibitor, angiotensin receptor blocker (ARB), diuretics, or an angio-tensin receptor-neprilysin inhibitor. Patients with more severe symptoms should be evaluated for placement of an implantable cardioverter-defibrillator, and may require cardiac transplantation in refractory cases.1,8
Figure 1. Stages of Heart Failure and Treatment
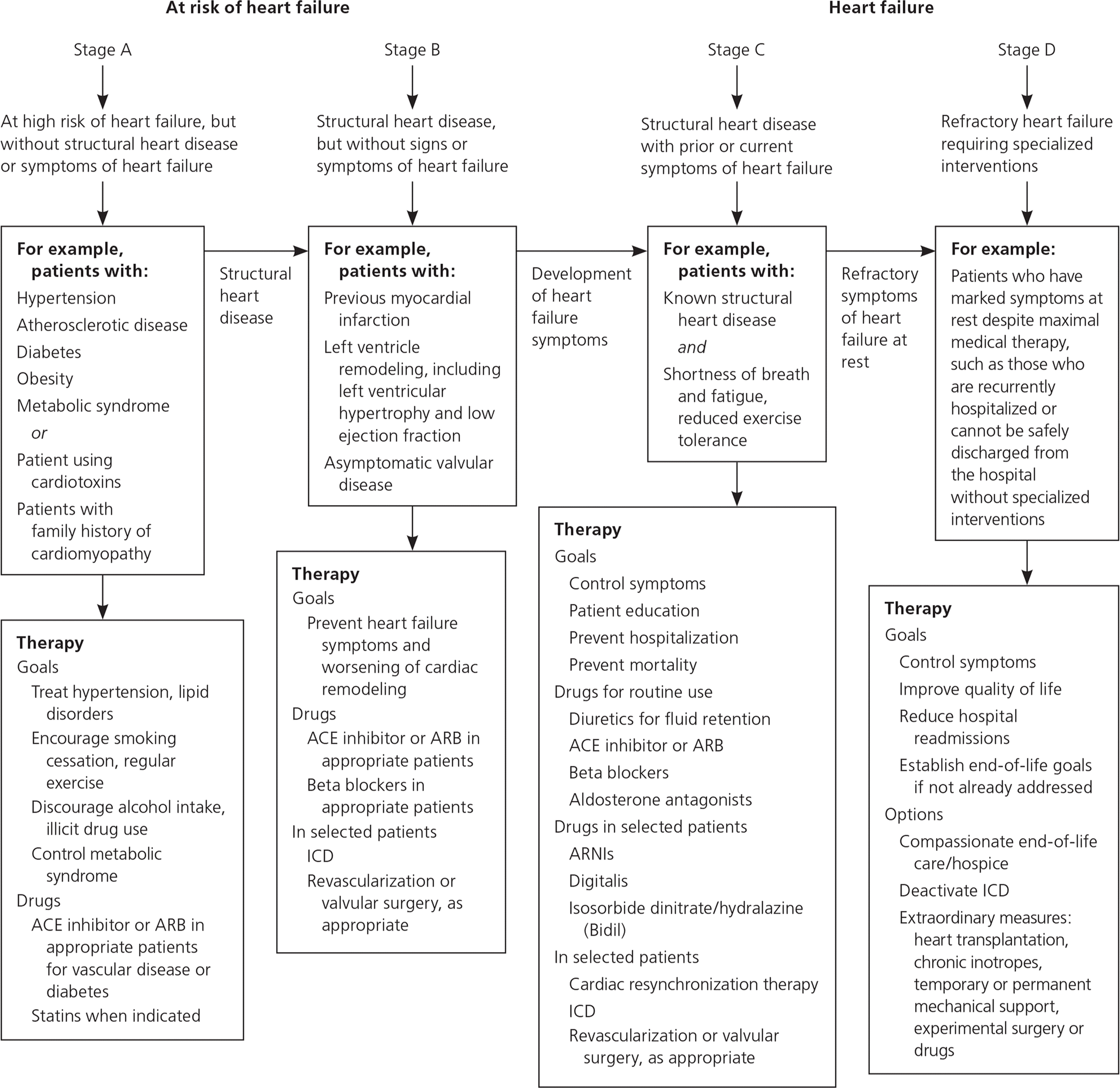
American College of Cardiology/American Heart Association heart failure guidelines. (ACE = angiotensin-converting enzyme; ARB = angiotensin receptor blocker; ARNI = angiotensin receptor-neprilysin inhibitor; ICD = implantable cardioverter-defibrillator.)
Adapted with permission from Hunt SA, Abraham WT, Chin MH, et al.; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; American College of Chest Physicians; International Society for Heart and Lung Transplantation; Heart Rhythm Society. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation. 2005;112(12):e154–235, with additional information from references 5 and 6.
Primary Cardiomyopathies
GENETIC ETIOLOGIES
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is the most common primary cardiomyopathy, with a prevalence of 1:500 persons.8 It is defined as left ventricular hypertrophy without chamber dilation and is caused by autosomal dominant mutations of genes that code for sarcomere proteins.1,9,10 Septal thickening predominates and may cause left ventricular outflow tract obstruction or mitral valve dysfunction.11 Phenotypic expression is variable, and some persons with HCM have a normal life expectancy with minimal or no disability.12
Many patients with HCM are asymptomatic and are diagnosed during family screening, by auscultation of a murmur, or incidentally after an abnormal result on electrocardiography. Presenting signs and symptoms most characteristic of HCM include atypical chest pain (which may be associated with meals, dehydration, or exertion) and sudden cardiac death.11,13 Patients who are diagnosed with HCM may have a family history of unexplained sudden cardiac death. On examination, physicians may hear a systolic murmur that increases in intensity during Valsalva maneuvers. Additionally, electrocardiography findings often show left ventricular hypertrophy and Q waves, and echocardiography results often show hypertrophy of the left ventricle coupled with reduction in ventricular chamber volume.12
HCM should be managed according to the 2011 guidelines from the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines.12 The main goals of therapy are to decrease exertional dyspnea and chest pain and prevent sudden cardiac death. Beta blockers are the initial therapy in patients with symptomatic HCM. Nondihydropyridine calcium channel blockers such as verapamil can be used if beta blockers are not well tolerated.13 All patients with HCM should undergo risk stratification for sudden cardiac death and be evaluated for placement of an implantable cardioverter-defibrillator12 (eFigure A). Additionally, these devices are recommended for secondary prevention of sudden cardiac death when there is any personal history of ventricular fibrillation or sustained ventricular tachycardia.1,12,14 Surgical myomectomy is recommended for end-stage refractory heart failure with left ventricular outflow obstruction unresponsive to medical therapy. Alcohol septal ablation, a minimally invasive procedure in which alcohol is injected via the septal artery to obliterate obstructing muscle tissue, is also an option if myomectomy is contraindicated (e.g., in patients with a very high surgical risk).15–17 In rare cases, heart transplantation may be considered for severe systolic symptoms.18
eFigure A. Hypertrophic Cardiomyopathy
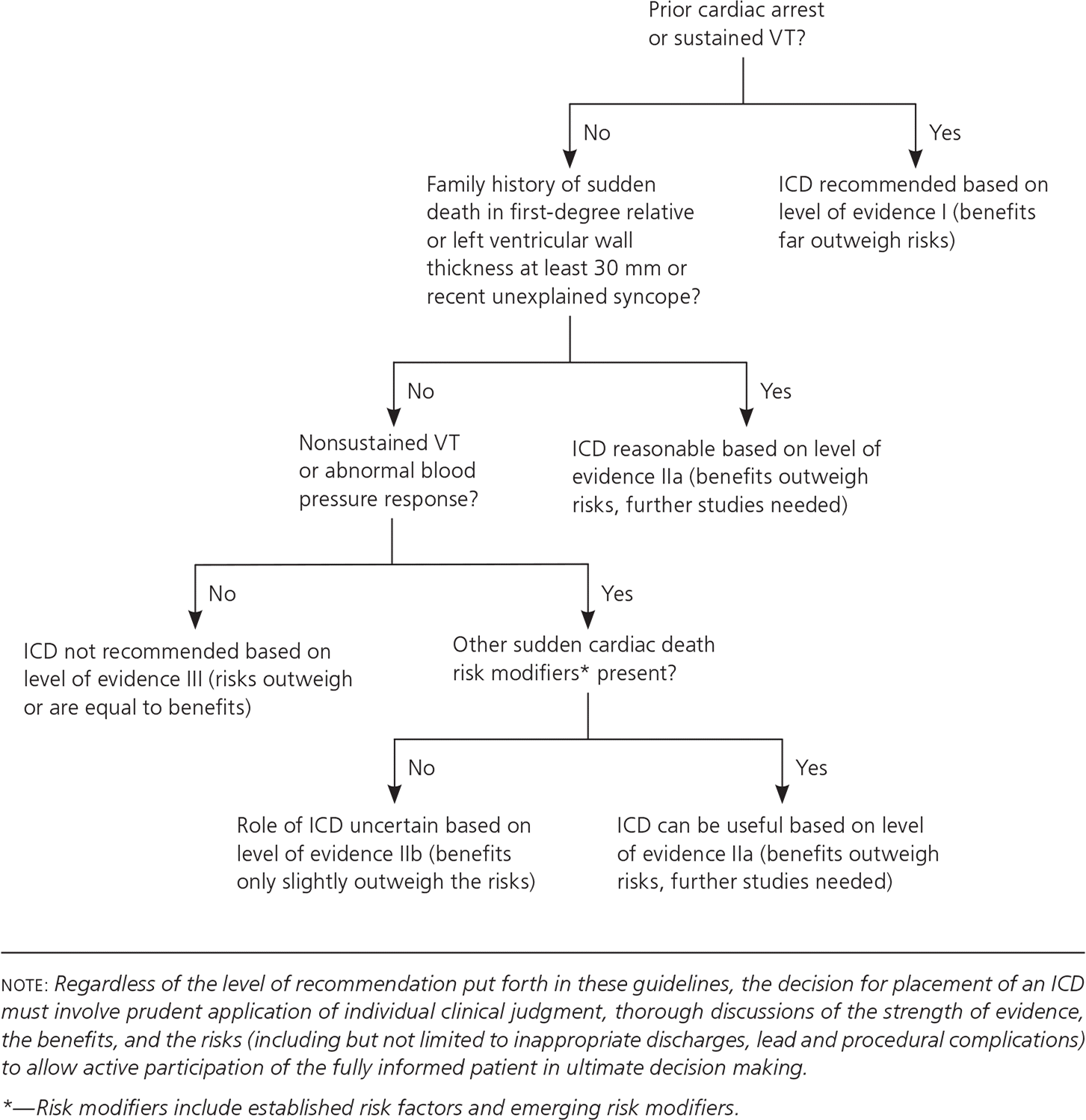
Evaluation for implantable cardioverter-defibrillator in hypertrophic cardiomyopathy. (ICD = implantable cardioverter-defibrillator; VT = ventricular tachycardia.)
Adapted with permission from Gersh BJ, Maron BJ, Bonow RO, et al.; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Failure Society of America; Heart Rhythm Society; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124(24):e783–e831.
Arrhythmogenic Right Ventricular Cardiomyopathy
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C), which has a prevalence of 1:1,000 to 5,000, is an inherited disease of desmosomal proteins that is characterized by fibrofatty infiltration of healthy myocardium.19,20 This process leads to thinning and ballooning of the ventricular wall, typically in the right ventricle.21 ARVD/C most commonly presents in the fourth decade of life with symptoms such as palpitations, syncope, and, occasionally, sudden cardiac death.19,22 Approximately one-half of cases are familial.19 Characteristic features on electrocardiography include inverted T waves and epsilon waves in the right precordial leads. Cardiac imaging may reveal right ventricular abnormalities, including aneurysms, segmental dilation, and reduced ejection fraction.22 Patients with ARVD/C are at increased risk of sudden cardiac death and should refrain from participating in competitive and endurance sports.23 Therapy is aimed at reducing arrhythmia and preventing sudden cardiac death and may include beta blockers, antiarrhythmic drugs, catheter ablation, implantable cardioverter-defibrillator placement, and heart transplantation.23
Left Ventricular Noncompaction
An uncommon and recently identified congenital cardiomyopathy is left ventricular noncompaction, a condition of embryonic origin that interferes with the development of mature heart muscle.1 Its exact prevalence is difficult to determine based on current data but has been estimated at less than 1% of the general population.24 The disease is defined by significant trabeculation of the myocardium, in addition to development of intertrabecular recesses in the left ventricle. These abnormalities lead to left ventricle dysfunction and, ultimately, to heart failure, arrhythmias, thromboembolic disease, and sudden cardiac death.25 Diagnosis is often made using imaging studies, typically echocardiography, although cardiac magnetic resonance imaging is recommended for confirmation.25 Standard heart failure treatment recommendations apply.24 In addition, patients with a history of atrial fibrillation, impaired systolic function, systemic embolism, or evidence of intracardiac thrombi should be treated with oral anticoagulants.1,24,25
MIXED ETIOLOGIES
Dilated Cardiomyopathy
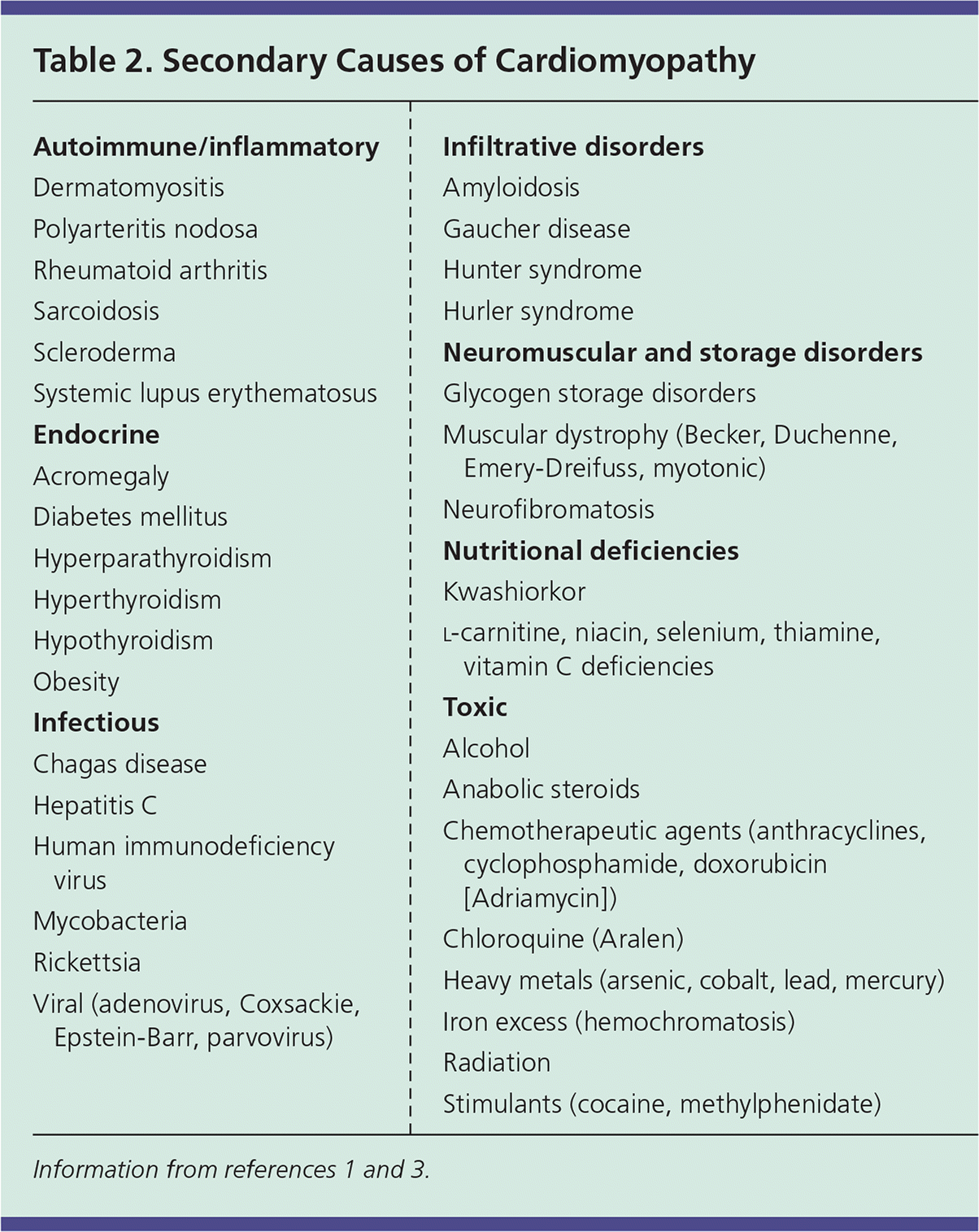
Dilated cardiomyopathy (DCM) has a prevalence of 1:2,500 and is the leading indication for heart transplantation.1 DCM is defined by enlargement of ventricles, normal left ventricular wall thickness, and systolic dysfunction.1 Approximately 25% to 35% of cases are familial, with such instances being primarily inherited in an autosomal dominant pattern.3 DCM may also result from a host of environmental, infectious, and systemic factors, as described in Table 2.1,3
Table 2. Secondary Causes of Cardiomyopathy
| Autoimmune/inflammatory |
| Dermatomyositis |
| Polyarteritis nodosa |
| Rheumatoid arthritis |
| Sarcoidosis |
| Scleroderma |
| Systemic lupus erythematosus |
| Endocrine |
| Acromegaly |
| Diabetes mellitus |
| Hyperparathyroidism |
| Hyperthyroidism |
| Hypothyroidism |
| Obesity |
| Infectious |
| Chagas disease |
| Hepatitis C |
| Human immunodeficiency virus |
| Mycobacteria |
| Rickettsia |
| Viral (adenovirus, Coxsackie, Epstein-Barr, parvovirus) |
| Infiltrative disorders |
| Amyloidosis |
| Gaucher disease |
| Hunter syndrome |
| Hurler syndrome |
| Neuromuscular and storage disorders |
| Glycogen storage disorders |
| Muscular dystrophy (Becker, Duchenne, Emery-Dreifuss, myotonic) |
| Neurofibromatosis |
| Nutritional deficiencies |
| Kwashiorkor |
| Toxic |
| Alcohol |
| Anabolic steroids |
| Chemotherapeutic agents (anthracyclines, cyclophosphamide, doxorubicin [Adriamycin]) |
| Chloroquine (Aralen) |
| Heavy metals (arsenic, cobalt, lead, mercury) |
| Iron excess (hemochromatosis) |
| Radiation |
| Stimulants (cocaine, methylphenidate) |
DCM can occur at any age, but is most common in patients 40 to 59 years of age.1,3 Symptoms characteristic of DCM include arrhythmias and thromboembolic events.26 Electrocardiography findings vary and may include isolated T wave changes, septal Q waves, bundle branch blocks, tachyarrhythmias, or normal results.3 Diagnosis is confirmed with echocardiography. Most patients are symptomatic at the time of diagnosis, but asymptomatic patients may be identified through screening of family members of affected patients.27 Treatment is guided by current evidence-based guidelines for heart failure1,5,6 (Figure 15–7 ). ACE inhibitors and ARBs have been shown to provide significant mortality benefit in patients with heart failure with reduced ejection fraction.28,29 Recent evidence further supports the use of sacubitril/valsartan [Entresto], an angiotensin receptor-neprilysin inhibitor, in place of an ACE inhibitor or ARB in patients with New York Heart Association class II or III heart failure with reduced ejection fraction.30 Beta blockade is also recommended in patients with heart failure with reduced ejection fraction.31–33
Restrictive Cardiomyopathy
Restrictive cardiomyopathy is the least common of the major cardiomyopathies, representing 2% to 5% of cases.34,35 The restrictive category includes many underlying etiologies and is defined by physiologic function rather than anatomy. The pattern of impaired ventricular filling with normal systolic function is typical, resulting from increased myocardial stiffness. Restrictive cardiomyopathy may be primary or secondary, with amyloidosis, sarcoidosis, radiation therapy, and scleroderma included among the common causes4 (Table 21,3 ).
Restrictive cardiomyopathy may present with signs of right-sided heart failure, such as ascites or peripheral edema. Examination may reveal elevated jugular venous pressure before the development of pulmonary edema. Chest radiography can detect pulmonary vascular congestion with a normal cardiac silhouette.34 Electrocardiography may show diffuse reduced voltage or a prolonged PR interval, and echocardiography may reveal biatrial enlargement and diastolic dysfunction, although left ventricular diastolic volume, wall thickness, and systolic function typically appear normal.4 Specific treatment options are limited and focus on addressing the underlying process. Symptomatic interventions include control of volume overload with diuretics or aldosterone antagonists and evaluation for atrioventricular block, with pacemaker insertion as indicated.36
ACQUIRED ETIOLOGIES
Peripartum Cardiomyopathy
Peripartum cardiomyopathy is defined as left ventricular systolic dysfunction at the end of pregnancy or in the months following delivery.37 Most patients present in the first month postpartum, although the condition may develop as early as the second trimester and as late as four months postpartum.38 Its incidence in the United States is unclear but estimated at 1:1,000 to 4,000 live births.38 Peripartum cardiomyopathy is associated with increasing age, black race, preeclampsia, hypertension, peripartum cardiomyopathy in a prior pregnancy, and multiple gestations.38 Presentation and physical examination findings are consistent with heart failure. Symptoms such as fatigue, edema, and dyspnea on exertion can be confused with more common pregnancy complications such as preeclampsia, and diagnosis of cardiomyopathy may be delayed.38,39 Electrocardiography findings are nonspecific, often showing only sinus tachycardia. Common echocardiography findings include left ventricular dilation, left ventricular systolic dysfunction, and pulmonary hypertension.38 Treatment follows standard heart failure therapy, with appropriate considerations for patients who are still pregnant. Therefore, ACE inhibitors and ARBs should be avoided in pregnant patients, and physicians should take care to avoid hypotension and reduced uterine perfusion when using diuretic therapy.38 Most women with peripartum cardiomyopathy recover left ventricular function. Long-term mortality rates have not been well documented but range from 11% to 16% in separate studies.38
Takotsubo Cardiomyopathy
Takotsubo cardiomyopathy, also known as stress-induced cardiomyopathy or broken-heart syndrome, is defined as an abrupt onset of left ventricular dysfunction in response to severe emotional or physiologic stress.1 Postmenopausal women are most commonly affected. The exact prevalence is difficult to determine but has been estimated at 0.02% of hospitalized patients, and it is possible that Takotsubo cardiomyopathy accounts for 1% to 2% of admissions for acute coronary syndrome.40,41 It often presents with angina, and typical ischemic changes may be seen with electrocardiography. A unique pattern of apical ballooning of the left ventricle is usually exhibited on echocardiography. Laboratory abnormalities may include elevated cardiac enzymes.42 Because its presentation closely mirrors that of acute coronary syndrome, Takotsubo cardiomyopathy initially should be treated in the same way. Acute complications, such as shock or heart failure, should be managed appropriately. Stable patients may be treated with diuretics, ACE inhibitors or ARBs, and beta blockers.42 Anticoagulants should be provided to patients with loss of wall motion in the left ventricular apex.42 Symptoms and abnormalities typically reverse within one month, and treatments may be withdrawn accordingly.5,42
Secondary Cardiomyopathies
Heart muscle disease resulting from an extracardiovascular cause is known as secondary cardiomyopathy. Although some etiologies are associated with specific disease patterns (e.g., alcohol use leading to dilated morphology, amyloidosis leading to a restrictive physiology), the expression of pathology caused by systemic disease is variable. Secondary causes can be grouped into several categories including endocrine, infectious, toxic, autoimmune, nutritional, and neuromuscular (Table 21,3 ). Evaluation and management are aimed primarily at the underlying disease process, removing offending agents, and treatment of the symptoms of heart failure.1
Screening
An autosomal dominant pattern of inheritance in HCM has been recognized for decades. Researchers identified mutations in genes coding for sarcomere proteins. Since then, more than 1,400 mutations among 13 genes have been identified as causing a heterogeneous phenotypic expression and course of illness. Patients with more than one mutation have an earlier onset of disease and a more severe course of illness. However, 40% to 50% of patients with HCM do not have a currently identified genetic mutation.9,43 Other inherited cardiomyopathies, such as ARVD/C and left ventricular noncompaction, have less robust genetic data to support diagnosis.44 Dilated and restrictive cardiomyopathies may have familial components, and genetic testing can lead to early diagnosis in family members of affected persons.27,44
Because of the preponderance of genetic influences on cardiomyopathy, it is generally recommended that patients with any cardiomyopathy be referred for genetic counseling, especially if the treating physician is not familiar with current guidelines.44 Early diagnosis may help guide treatment decisions, improve quality of life, or even prolong life expectancy.10,44–46 Further, first-degree relatives of patients with cardiomyopathy should be considered for clinical and genetic screening. Clinical screening may include complete history and physical examination, electrocardiography, and echocardiography. Guidelines regarding optimal screening intervals and referral to genetic counseling are emerging.44
Activity Restrictions
HCM is the most common cause of sudden cardiac death in athletes, accounting for about one-third of cases. This devastating complication is more prevalent in male athletes and nonwhites.47 Current guidance holds that patients with phenotypic HCM should not participate in intense competitive sports but may participate in low-intensity activities.48 The restrictions on genotype-positive, phenotype-negative competitive athletes are controversial. The European Society of Cardiology suggests full restrictions, whereas the United States 36th Bethesda Conference guidelines find insufficient evidence to exclude these persons from competitive sports.48,49
Data Sources: We searched the Cochrane Database of Systematic Reviews, PubMed, Clinical Key, National Guideline Clearinghouse, Dynamed, and Essential Evidence using the key words cardiomyopathy, hypertrophic cardiomyopathy, dilated cardiomyopathy, restrictive cardiomyopathy, heart failure, and congestive heart failure. Some sources were revisited. Search dates: March, June, and July 2016 and July 2017.
