Although the prevalence of muscle weakness in the general population is uncertain, it occurs in about 5% of U.S. adults 60 years and older. Determining the cause of muscle weakness can be challenging. True muscle weakness must first be differentiated from subjective fatigue or pain-related motor impairment with normal motor strength. Muscle weakness should then be graded objectively using a formal tool such as the Medical Research Council Manual Muscle Testing scale. The differential diagnosis of true muscle weakness is extensive, including neurologic, rheumatologic, endocrine, genetic, medication- or toxin-related, and infectious etiologies. A stepwise approach to narrowing this differential diagnosis relies on the history and physical examination combined with knowledge of the potential etiologies. Frailty and sarcopenia are clinical syndromes occurring in older people that can present with generalized weakness. Asymmetric weakness is more common in neurologic conditions, whereas pain is more common in neuropathies or radiculopathies. Identifying abnormal findings, such as Chvostek sign, Babinski reflex, hoarse voice, and muscle atrophy, will narrow the possible diagnoses. Laboratory testing, including electrolyte, thyroid-stimulating hormone, and creatine kinase measurements, may also be helpful. Magnetic resonance imaging is indicated if there is concern for acute neurologic conditions, such as stroke or cauda equina syndrome, and may also guide muscle biopsy. Electromyography is indicated when certain diagnoses are being considered, such as amyotrophic lateral sclerosis, myasthenia gravis, neuropathy, and radiculopathy, and may also guide biopsy. If the etiology remains unclear, specialist consultation or muscle biopsy may be necessary to reach a diagnosis.
Given its broad differential diagnosis, muscle weakness can be challenging to evaluate in primary care practice. Although its prevalence in the general population is not well described, muscle weakness occurs in 5% of U.S. adults 60 years and older.1 Physicians must distinguish true muscle weakness from subjective fatigue or pain-related motor impairment with normal motor strength. This requires a history and physical examination, which guide laboratory testing, imaging, electrodiagnostic testing, and muscle biopsy.2–5
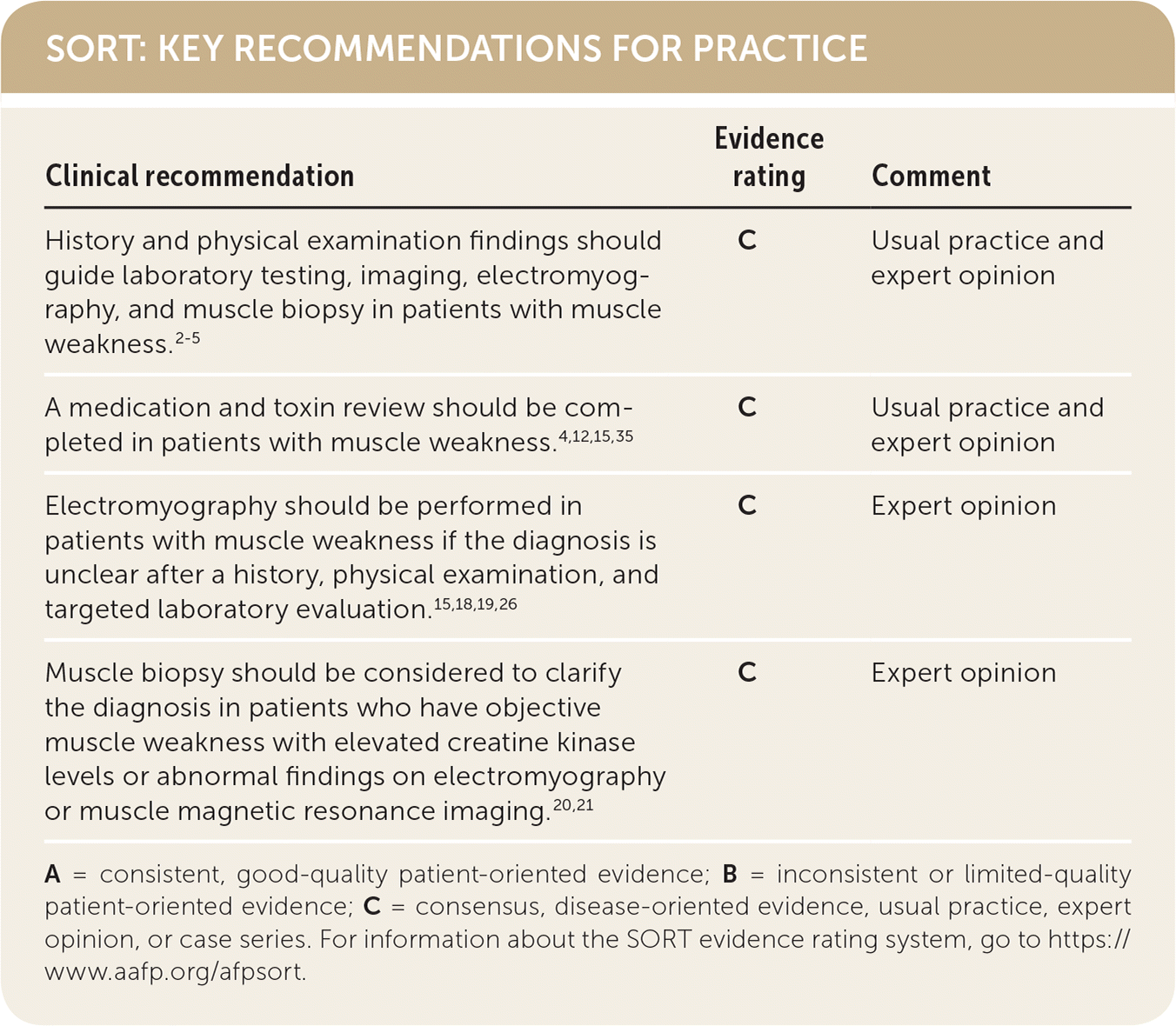
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | Comment |
|---|---|---|
| History and physical examination findings should guide laboratory testing, imaging, electromyography, and muscle biopsy in patients with muscle weakness.2–5 | C | Usual practice and expert opinion |
| A medication andtoxin review should be completed in patients with muscle weakness.4,12,15,35 | C | Usual practice and expert opinion |
| Electromyography should be performed in patients with muscle weakness if the diagnosis is unclear after a history, physical examination, and targeted laboratory evaluation.15,18,19,26 | C | Expert opinion |
| Muscle biopsy should be considered to clarify the diagnosis in patients who have objective muscle weakness with elevated creatine kinase levels or abnormal findings on electromyography or muscle magnetic resonance imaging.20,21 | C | Expert opinion |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.
Etiology and Differential Diagnosis
The differential diagnosis of muscle weakness in adults is extensive because it can occur when pathology affects any level of the neuromuscular pathway (upper or lower motor neurons, neuromuscular junction, and muscle fibers). Table 1 lists selected causes of muscle weakness and their clinical findings.5–22
TABLE 1. Selected Causes of Muscle Weakness in Adults
| Cause | Weakness | Upper or lower motor neuron signs | Pattern of onset | Systemic symptoms and findings | Laboratory or imaging | Creatine kinase level | Electromyography | Muscle biopsy |
|---|---|---|---|---|---|---|---|---|
| Neurologic | ||||||||
| Amyotrophic lateral sclerosis | Variable, typically starts unilaterally in one peripheral region of the body and progresses into others; may present with impairment of the ninth to 12th cranial nerves (bulbar palsy) | Both | Gradual | Muscle cramps, muscle atrophy (often in the hands); fasciculations, including in the tongue; spasticity; may exhibit pseudobulbar affect | MRI of brain and spinal cord may help rule out other causes of weakness | Normal or elevated | Reduced CMAP amplitude with normal sensory nerve amplitude; fibrillation potentials,* positive sharp waves | Not typically performed |
| Cerebral lesion (stroke, hemorrhage, mass) | Variable depending on lesion location, often unilateral | Upper | Acute (stroke), subacute or gradual (mass, subdural hematoma) | May present with headache, altered cognition, or altered sensorium | MRI may reveal etiology | Not typically obtained | Not typically performed | Not typically performed |
| Compressive neuropathy of the peripheral nerve, (e.g., carpal tunnel syndrome, lumbar radiculopathy) | Variable, unilateral | Lower | Variable | Pain, paresthesia; spinal nerve root compression may cause radicular symptoms | Ultrasonography may reveal nerve entrapment | Not typically obtained | Fibrillation potentials,* positive sharp waves; reduced MUAP amplitude distal to offending lesion | Not typically performed |
| Guillain-Barré syndrome | Symmetric, bilateral; affects distal and proximal muscles; may progress to include respiratory and bulbar involvement | Lower | Acute | History of recent Campylobacter jejuni, Epstein-Barr virus, cytomegalovirus, or mycoplasma infection; recent vaccination; may be associated with back pain and symmetric sensory changes (e.g., paresthesias) | Anti-GM1 antibody, anti-GD1a antibody; cerebrospinal fluid analysis may show increased protein and normal leukocytes after the first week of illness | Not typically obtained | May be normal in first week of illness; later shows patchy demyelination findings of temporal dispersion of CMAPs; slowed motor conduction velocity and/or conduction block | Not typically performed |
| Lambert-Eaton syndrome | Proximal, caudocranial progression; initially improves with repetition; diminished tendon reflexes | Lower | Variable | Dysautonomia, impotence, hypotension, xerostomia; often develops as a paraneoplastic syndrome associated with small cell lung cancer but may be idiopathic | Anti–voltage-gated calcium channel antibody | Normal | Low CMAP amplitude with rest, improvement postexercise; fibrillation potentials* | Not typically performed |
| Motor neuropathy | Distal, eventually proximal | Lower | Gradual | May have accompanying sensory neuropathy with paresthesia or numbness; toe deformities, foot ulcers; impaired balance | Various genetic tests depending on suspected etiology; elevated serum glucose or A1C level if caused by diabetic neuropathy; muscle atrophy on MRI | Not typically obtained | Low CMAP amplitude in lower extremities; demyelination signs in upper extremities | Not typically performed |
| Multiple sclerosis | Variable depending on location, often unilateral | Upper | Variable | May present with bladder dysfunction, cognitive impairment, sensory changes, monocular vision loss due to optic neuritis, cerebellar ataxia | MRI may show characteristic white matter lesions of the brain or spinal cord | Not typically obtained | Not typically performed | Not typically performed |
| Myasthenia gravis | Proximal, cephalocaudal progression; worsens with repetition (fatigability); oculomotor | Lower | Variable | Worsening of symptoms throughout the day | Anti–acetylcholine receptor antibody testing; anti–striated muscle antibody and anti–muscle specific tyrosine kinase antibody testing have less utility | Normal | Normal CMAP amplitude at rest, reduced CMAP amplitude with repetitive nerve stimulation; fibrillation potentials* | Not typically performed |
| Spinal cord lesion (stroke, hemorrhage, mass) | Variable depending on lesion location, may be unilateral or bilateral | Upper | Acute (stroke), gradual (mass) | Back pain, bowel or bladder incontinence | MRI may reveal etiology | Not typically obtained | Not typically performed | Not typically performed |
| Myositides | ||||||||
| Dermatomyositis | Proximal | — | Acute or subacute | Violaceous rash on the knuckles and extensor surfaces of the upper extremities (Gottron papules [Figure 2]), violaceous rash on the eyelids and periorbital edema (heliotrope rash), violaceous rash on the upper chest and neck (V-sign) or back (shawl sign); calcinosis; interstitial lung disease; disordered GI motility; dysphagia | Elevated myoglobin; positive for antinuclear antibodies; may be positive for myositis autoantibodies | Greater than 10 times the normal elevations | Active and chronic myopathic units | Inflammatory infiltrate with myopathic changes† and replacement by adipose and collagen |
| Inclusion body myositis | Distal, especially forearm and hand; later stage may involve proximal weakness | — | Gradual | Dysphagia (present in up to 80% of cases); extramuscular involvement not as common | Elevated myoglobin; antinuclear antibodies less common; myositis autoantibodies may be present | Elevated | Active and chronic myopathic units | Inflammatory infiltrate with vacuoles containing eosinophilic inclusions; ragged red or ragged blue fibers |
| Necrotizing myositis | Proximal, often profound | — | Acute | May be associated with viral illness, malignancy, or statin use; dysphagia | Anti–signal recognition particle antibody; anti–3-hydroxy-3-methylglutaryl coenzyme A reductase antibody | Markedly elevated (can be over 50 times normal limit) | Active myopathic units | Scattered necrosis, deposits of complement may be noted on capillaries‡ |
| Polymyositis | Proximal | — | Acute or subacute | Similar in presentation to dermatomyositis but without dermatologic signs; interstitial lung disease; disordered GI motility; dysphagia | Elevated myoglobin; antinuclear antibodies less common; myositis autoantibodies may be present | Greater than 10 times normal elevations | Active and chronic myopathic units | Inflammatory infiltrate with myopathic changes† and replacement by adipose and collagen |
| Endocrine | ||||||||
| Adrenal insufficiency (Addison disease) | Generalized | — | Variable | Hypotension, hypoglycemia, bronzing of the skin | Hyponatremia, hyperkalemia; adrenocorticotropic hormone assay and stimulation test | Normal | Myotonic discharges§ | Diminished glycogen content |
| Elevated thyroid hormone (hyperthyroidism) | Proximal, bulbar | — | Variable | Weight loss, tachycardia, increased perspiration, tremor | Elevated thyroxine (T4) and triiodothyronine (T3) hormones; thyroid-stimulating hormone variable depending on cause | Normal or elevated | Myopathic MUAPs‖ with or without fibrillation potentials* | Usually normal |
| Endogenous glucocorticoid excess (Cushing syndrome) | Proximal | — | Variable | Buffalo hump, purple striae, osteoporosis | Elevated urine-free cortisol, dexamethasone suppression, or corticotropin-releasing hormone stimulation tests | Normal or low | Myopathic MUAPs‖ | Selective atrophy of type II muscle fibers, absent necrosis |
| Low/absent thyroid hormone (hypothyroidism) | Proximal | — | Variable | Menorrhagia, bradycardia, goiter, delayed relaxation of deep tendon reflexes | Thyroid-stimulating hormone | Elevated | With or without myopathic MUAPs‖ and fibrillation potentials* | Myopathic changes,† glycogen accumulation |
| Parathyroid hormone (secondary hyperparathyroidism¶) | Proximal, lower extremity more than upper extremity | — | Variable | Usually has associated comorbidities (cardiovascular disease, diabetes mellitus); positive Chvostek or Trousseau signs | Hypocalcemia; uremia | Normal | Myopathic MUAPs‖ | Atrophy of type II muscle fibers;increased lipofuscin beneath cell membrane; calcium deposits in muscle |
| Genetic | ||||||||
| Becker muscular dystrophy | Hip, proximal leg and arm | — | Gradual | Intellectual disability, cardiomyopathy | Various specific molecular genetic tests are available | Elevated | Myopathic MUAPs‖ with fibrillation potentials* | Myopathic changes,† decreased and patchy staining of dystrophin |
| Glycogen and lipid storage diseases, mitochondrial diseases | Proximal | — | Variable | Variable; exercise intolerance and cardiomyopathy more common | Some glycogenoses associated with abnormal findings on forearm ischemic exercise testing** | Variable, may increase with exercise | Normal or myopathic MUAPs‖ with or without fibrillation potentials* | Myopathic changes† with glycogen deposits, lipid deposits, or ragged red fibers (for glycogen, lipid, or mitochondrial disease, respectively) |
| Limb-girdle muscular dystrophy | Variable; usually proximal limb, pelvic, and shoulder girdle muscles | — | Gradual | Variable, may have cardiac abnormalities | Various specific molecular genetic tests available | Variable, normal, or elevated | Myopathic MUAPs‖ with or without fibrillation potentials* | Myopathic changes,† may demonstrate absence of specific protein on immunohistochemical staining |
| Myotonic dystrophy type 1 | Distal greater than proximal, foot drop, oropharyngeal, temporal and masseter wasting, difficulty opening hand after tight grip | — | Gradual | Cardiac conduction abnormalities, balding, intellectual disability, gonadal atrophy, cataracts, insulin resistance, scoliosis | Molecular genetic testing may demonstrate unstable CTG trinucleotide repeat in the DMPK gene of chromosome 19 | Normal to minimally elevated | Myopathic MUAPs,‖ myotonic discharges* | Less necrosis and remodeling than in muscular dystrophy, atrophy of type I muscle fibers, ring fibers |
| Medications | ||||||||
| Fluoroquinolones | Distal | — | Acute or subacute | Tendon swelling or erythema, tendon rupture in severe cases | Ultrasonography or MRI may demonstrate tendon rupture | — | Not typically performed | Not typically performed |
| Glucocorticoids | Typically affects proximal and postural muscles | — | Acute, subacute, or gradual | Buffalo hump, purple striae | No reliable biomarkers; may have leukocytosis | Normal or low | Myopathic MUAPs‖ | Selective atrophy of type II muscle fibers, absent necrosis |
| Statin | Proximal (may be distal) | — | Acute or subacute | Myalgia often affecting bilateral large muscle groups, such as back, buttock, thigh, and shoulder girdle muscles; onset shortly after medication initiation; higher risk in females, older people, and those with lower muscle mass | None | Elevated | Myopathic MUAPs‖ | Myonecrosis in severe cases |
| Toxins | ||||||||
| Alcohol (myopathy or neuropathy) | Proximal (may be distal) | Alcoholic neuropathy: lower; alcoholic myopathy: — | Variable | Change in mental status, telangiectasia, peripheral neuropathy | Elevated transaminase and gamma-glutamyltransferase levels, anemia, decreased vitamin B levels12 | Normal to elevated | Normal | Myopathic changes,† selected atrophy of type II muscle fibers |
| Catabolic | ||||||||
| Sarcopenia and frailty | Variable, slow gait speed, diminished grip strength | — | Gradual onset in patients older than 65 years | Loss of muscle mass, cachexia | No specific biomarkers but may have low creatinine, decreased insulinlike growth factor 1, albumin, or vitamin D levels; may have elevated interleukin-6 | Normal or low | Not typically performed | Sample may be inadequate because of lack of muscle mass; fatty infiltration |
CMAP = compound muscle action potential; GI = gastrointestinal; MRI = magnetic resonance imaging; MUAP = motor unit action potential.
*—Fibrillation potentials represent spontaneous activity of single muscle fibers (not usually noted with normal muscle).
†—Myopathic changes are nonspecific and include atrophy, degeneration, and regeneration of muscle fibers.
‡—C3 and C4 are complement components.
§—Myotonic discharges are a type of prolonged burst of activity seen on insertion of the electromyography needle.
‖—Myopathic MUAPs are shorter in duration, lower in amplitude, and polyphasic compared with MUAPs from normal muscle.
¶—Secondary hyperparathyroidism usually caused by renal failure.
**—Forearm ischemic exercise testing evaluates the rise of ammonia and lactate in the forearm during exertion.
Adapted with permission from Saguil A. Evaluation of the patient with muscle weakness. Am Fam Physician. 2005;71(7):1330–1331, with additional information from references 5 and 7–22.
NEUROLOGIC
Upper motor neurons, which are located in the cerebral cortex, brainstem, and corticospinal tracts, may be injured from stroke, multiple sclerosis, and spinal cord or brain masses. Lower motor neurons, which are located in the anterior horn of the spinal cord, nerve roots, and peripheral nerves, may be damaged by infections, Guillain-Barré syndrome, hereditary sensorimotor disorders (e.g., Charcot-Marie-Tooth disease), compressive neuropathy (e.g., carpal tunnel syndrome), radiculopathy from intervertebral disk herniation, inflammation, spinal muscular atrophy type IV, or amyloid infiltration.2,23–25
Upper and lower motor neurons are involved in amyotrophic lateral sclerosis.24 The neuromuscular junction is the site of pathology in myasthenia gravis and Lambert-Eaton syndrome, which are rare autoimmune-mediated disorders.7,8
MYOSITIDES
Weakness is the predominant feature of primary muscle disorders, or myositides. Some of these disorders, such as dermatomyositis, polymyositis, and necrotizing myositis, present with acute or subacute proximal weakness and may be associated with malignancy or connective tissue disorders. Conversely, inclusion body myositis, which tends to occur in older adults, has a gradual course, affecting distal muscles early and progressing in an asymmetric pattern.9,10,26,27
ENDOCRINE
Some endocrine disorders can cause weakness. These include hyperthyroidism and hypothyroidism, adrenal insufficiency (Addison disease), endogenous glucocorticoid excess (Cushing syndrome), acromegaly, and primary hyperaldosteronism (Conn syndrome).3 Diabetic neuropathy may affect motor neurons, including the oculomotor nerve.28
ELECTROLYTE DISTURBANCES
Numerous electrolyte imbalances are linked to muscle weakness, including disturbances in calcium, phosphate, potassium, and sodium. Hypokalemia is particularly common and can be caused by medications, such as diuretics; Gitelman syndrome (hypokalemic periodic paralysis); renal tubular acidosis; primary hyperaldosteronism; thyrotoxicosis; or infections, such as dengue fever.3,4,29–31
GENETIC
Muscular dystrophy usually presents during childhood or adolescence, but certain subtypes (e.g., Becker muscular dystrophy, myotonic dystrophy) can present during adulthood.11 Disorders of lipid and glycogen metabolism and disorders of mitochondria can also cause weakness, myopathy, and rhabdomyolysis; these disorders may be exacerbated by exercise or febrile illnesses and rarely present in adults.32,33
MEDICATIONS
Numerous medications have been reported to cause muscle weakness (Table 24,12–14,34 ), including several commonly used medications. For example, statins are well known to cause myopathy and rhabdomyolysis, although the mechanism is unclear.12,13 Fluoroquinolones are linked to tendinopathies, rhabdomyolysis, and tendon ruptures.12 Glucocorticoids can cause a noninflammatory myopathy, primarily affecting postural muscles.12,14 A medication review is thus essential in the evaluation of muscle weakness.4,12,35
TABLE 2. Medications that Can Cause Muscle Weakness

| Amiodarone Antimicrotubular medications (colchicine, vincristine) Antiretroviral medications (nucleoside reverse transcriptase inhibitors, such as zidovudine [Retrovir], stavudine, and lamivudine [Epivir]; protease inhibitors, such as indinavir [Crixivan], saquinavir [Invirase], and ritonavir [Norvir]) Any medication causing hypocalcemia/hypercalcemia or hypokalemia/hyperkalemia Fibrates* Fluoroquinolones Gemfibrozil (Lopid) Glucocorticoids (systemic) HMG-CoA reductase inhibitors (statins)†Hydroxychloroquine (Plaquenil) Hydroxyurea Interferon alfa* Local anesthetics Omeprazole (Prilosec)* Penicillamine (Cuprimine)* Phenytoin (Dilantin)* Silicone gel (ruptured breast implant)* |
CYP3A4 = cytochrome P450 3A4.
*—Can cause weakness via drug-induced dermatomyositis.
†—Concurrent use of statins and CYP3A4 inhibitors increases the likelihood of statin toxicity. Select CYP3A4 inhibitors include amiodarone, calcium channel blockers (e.g., amlodipine [Norvasc], diltiazem, verapamil), certain anti-HIV agents (e.g., ritonavir, saquinavir), certain antidepressants (e.g., fluoxetine [Prozac], fluvoxamine), certain antifungals (e.g., fluconazole [Diflucan], itraconazole [Sporanox], ketoconazole, miconazole), tamoxifen, and grapefruit juice.
TOXINS
Exposure to heavy metals, such as arsenic, lead, thallium, and mercury, can cause motor neuropathy; therefore, a toxin review should be completed in patients with muscle weakness.4,35 Organophosphate poisoning causes weakness at the level of the neuromuscular junction, with severe cholinergic effects (toxidrome). Alcohol and certain recreational drugs (e.g., “glue sniffing,” cocaine, amphetamines, opioids) can also cause muscle weakness. Alcohol is neurotoxic and myotoxic at high doses and can cause acute muscle weakness in individuals who binge drink.15
Envenomation, including from tick bites (tick paralysis) and certain venomous snake bites, may cause weakness. Exposure to Clostridium botulinum toxin from eating contaminated or undercooked food causes acute paralysis by blocking release of acetylcholine into the neuromuscular junction.4,11,35
INFECTIONS
Certain infections are linked to muscle weakness. These include West Nile virus infection, HIV infection, Lyme disease, diphtheria, dengue fever, neurocysticercosis, trichinosis, Chagas disease, rabies, botulism, herpes zoster, cytomegalovirus infection, hepatitis C, and herpes simplex virus 1 infection. Lyme disease, herpes zoster, and potentially herpes simplex virus 1 infection can cause facial nerve palsy (Bell palsy). Poliomyelitis was historically a significant cause of flaccid paralysis but now rarely occurs because of worldwide vaccination efforts.3,23,24,29,30,36,37
SARCOPENIA AND FRAILTY
When evaluating generalized weakness in older adults, sarcopenia and frailty should be considered in the differential diagnosis. Sarcopenia and frailty are common multifactorial syndromes that typically do not occur in younger people. They are associated with increased morbidity, disability, institutionalization, and mortality. Sarcopenia is thought to be catabolic age-related loss of muscle mass and strength and can be evaluated by measuring grip strength, gait speed, and muscle mass.38,39 Frailty, which often coexists with sarcopenia, can be defined as the presence of three or more of the following: grip weakness, slow gait speed, unintentional weight loss, subjective exhaustion, and low physical activity.40
History
Physicians should inquire about onset, duration, and progression of symptoms. Acute onset of weakness (hours to days) should prompt timely evaluation, because it may indicate vascular, infectious, inflammatory, metabolic, or toxin-mediated disorders. Subacute presentation (days to weeks) may suggest electrolyte, inflammatory, or rheumatologic disorders. More gradual progressive weakness often suggests neurologic, genetic, or metabolic disorders or inclusion body myositis.2,9,11,29
The history should be guided by knowledge of the potential diagnoses and proceed in a stepwise approach (Figure 12–5,7–24,26–33,35–38,41–44 ). Specific elements of the history may provide diagnostic clues (Table 32–4,7–11,13,22–24,26–28,31,35,43,44 ).
FIGURE 1.
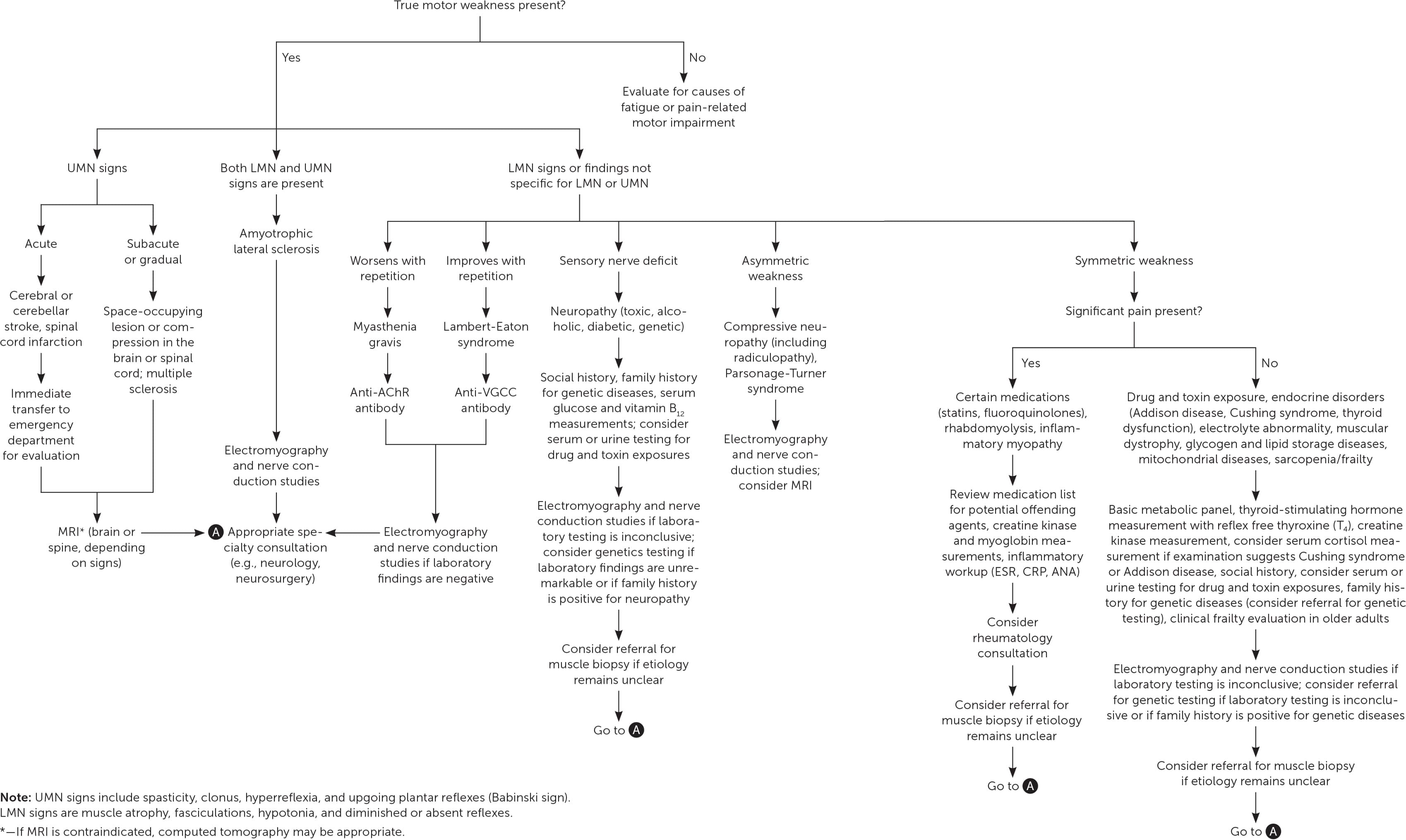
Evaluation of muscle weakness in adults.
AChR = anti–acetylcholine receptor; ANA = antinuclear antibodies; CRP = C-reactive protein; ESR = erythrocyte sedimentation rate; LMN = lower motor neuron; MRI = magnetic resonance imaging; UMN = upper motor neuron; VGCC = voltage-gated calcium channel.
Information from references 2–5, 7–24, 26–33, 35–38, and 41–44.
TABLE 3. Diagnostic Clues in the Evaluation of Muscle Weakness

| Key findings | Suggested diagnoses |
|---|---|
| History | |
| Diplopia, blurry vision | Myasthenia gravis (intermittent), diabetic neuropathy (permanent) |
| Dysphagia | Inflammatory myopathy, motor neuron disease, myasthenia gravis, muscular dystrophy, botulism |
| Facial nerve paralysis (Bell palsy) | Lyme disease, herpes zoster (Ramsay Hunt syndrome), herpes simplex virus 1 infection |
| Fevers, arthralgias, rash | Inflammatory or infectious myopathy |
| Monocular vision loss, bladder dysfunction | Multiple sclerosis |
| Pain | Nerve compression, rhabdomyolysis, inflammatory myopathy, inflammatory neuropathy (e.g., Parsonage-Turner syndrome), muscle damage due to toxins or medications (e.g., statins, fluoroquinolones) |
| Paresthesia | Hereditary sensorimotor disorders (e.g., Charcot-Marie-Tooth disease), nerve compression, nerve damage due to toxins, multiple sclerosis |
| Raynaud phenomenon | Inflammatory myopathy |
| Tendon rupture | Fluoroquinolone use |
| Physical examination | |
| Cardiac arrhythmias | Inflammatory myopathy, genetic disorders, electrolyte imbalances |
| Cholinergic toxidrome | Organophosphate poisoning |
| Chvostek sign,* Trousseau sign,† tetany | Hypocalcemia |
| Cushingoid appearance (buffalo hump, purple striae) | Glucocorticoid excess (endogenous [Cushing syndrome] vs. exogenous [long-term glucocorticoid use]) |
| Fatigable weakness, ptosis | Myasthenia gravis |
| Hyperreflexia | Upper motor neuron lesion, hyperthyroidism, hypercalcemia |
| Hyporeflexia | Hypothyroidism, lower motor neuron lesion |
| Muscle atrophy | Lower motor neuron pathology, frailty/sarcopenia |
| Skin bronzing | Hypoaldosteronism (Addison disease) |
| Third cranial nerve (oculomotor nerve) palsy | Diabetic neuropathy |
| Violaceous rash on the knuckles and extensor surfaces of upper extremities (Gottron papules [Figure 2]), violaceous rash on the eyelids and periorbital edema (heliotrope rash), violaceous rash on the upper chest and neck (V-sign) or back (shawl sign) | Dermatomyositis |
*—Tapping the facial nerve anterior to the earlobe and inferior to the zygomatic arch elicits ipsilateral facial nerve spasm.
†—Inflating a brachial blood pressure cuff induces distal carpopedal spasm.
Information from references 2–4, 7–11, 13, 22–24, 26–28, 31, 35, 43, and 44.
In particular, identifying the distribution of weakness helps narrow the broad differential diagnosis. Patients with proximal (limb-girdle) weakness may have difficulty rising from a chair, climbing stairs, or lifting their arms overhead to perform activities of daily living (e.g., combing hair).2,26 Proximal weakness is more common in myopathies. Patients with distal weakness may have difficulty opening jars or may drop objects or have foot drop. Distal weakness is more typically caused by certain toxins (e.g., organophosphates), hereditary motor neuropathy, or early inclusion body myositis.3,9,16,41 Simultaneous proximal and distal weakness may result from inclusion body myositis or Guillain-Barré syndrome.
Asymmetric weakness is typically neurologic and may be due to central nervous system lesions or peripheral nerve inflammation or compression. The physician should also inquire about the presence of oropharyngeal symptoms (e.g., dysphagia, dysarthria), vision loss, or diplopia, which can be indicative of inflammatory myopathy, multiple sclerosis, or myasthenia gravis, respectively.2,9–11,27
The presence of accompanying sensory symptoms suggests a neuropathic etiology from intrinsic or extrinsic factors. For example, pain in the same distribution as the weakness suggests nerve compression, inflammatory neuropathy or myopathy, or muscle damage due to toxins or medications. History of fevers, arthralgias, rash, or Raynaud phenomenon suggests inflammatory etiologies. Fever also suggests infectious etiologies. All types of inflammatory myopathy can present with pain and muscle tenderness.3,10,26
Patients with myasthenia gravis often present with intermittent blurry vision or diplopia and less commonly with more generalized weakness. In addition, symptoms of myasthenia gravis often fluctuate, worsen throughout the day, and may be exacerbated by elevated temperatures.7
A family history may help identify hereditary causes of muscle weakness.2,11,17 A medication review and screening for substance use should always be performed in patients with muscle weakness.15,35
Physical Examination
Physical examination is crucial to diagnosing the cause of muscle weakness. Weakness should be formally graded and documented with a tool such as the Medical Research Council Manual Muscle Testing scale (Table 445 ).
TABLE 4. Medical Research Council Manual Muscle Testing Scale for Assessing Muscle Strength

| 5: Muscle movement/activation against examiner's full resistance |
| 4: Muscle strength is reduced but movement/activation against some resistance is possible |
| 3: Muscle movement/activation against gravity (e.g., raising leg or arm) but not against resistance |
| 2: Muscle movement/activation only when not moving against gravity (e.g., not lifting leg or raising arm) |
| 1: Trace muscle movement/activation (e.g., a twitch) |
| 0: No muscle movement/activation |
Note: The patient's strength is graded from 0 (no movement) to 5 (full strength).
Adapted with permission from Naqvi U, Sherman AI. Muscle strength grading. StatPearls. February 17, 2019. https://www.ncbi.nlm.nih.gov/books/NBK436008/
NEUROLOGIC EXAMINATION
A neurologic examination should be performed in patients with muscle weakness to observe signs of upper motor neuron vs. lower motor neuron pathology. Upper motor neuron findings include spasticity (e.g., spastic gait), hyperreflexia, upgoing-toe plantar reflexes (positive Babinski reflex), dysarthria, clonus, and poor coordination. Lower motor neuron findings include diminished or absent reflexes, hypotonia, muscle atrophy (e.g., hoarse voice from laryngeal muscle atrophy), and fasciculations. Visual inspection of the muscles with attention to bulk, involuntary movements, and symmetry is also important.
Hyperreflexia can occur with upper motor neuron lesions, hyperthyroidism, and hypercalcemia. Hypocalcemia is suggested with tetany, Chvostek sign (tapping the facial nerve anterior to the earlobe and inferior to the zygomatic arch elicits ipsilateral facial nerve spasm), and Trousseau sign (inflation of a brachial blood pressure cuff induces distal carpopedal spasm). Up to 94% of patients with hypocalcemia present with Trousseau sign, although Chvostek sign is neither sensitive nor specific for hypocalcemia.43,44
Cranial nerve examination can reveal facial nerve weakness suggesting Lyme disease, herpes simplex virus 1 infection, or herpes zoster (Ramsay Hunt syndrome).37 Ptosis or diplopia may suggest myasthenia gravis. Motor strength worsens with repetition in patients with myasthenia gravis, whereas strength and reflexes may transiently improve after repetitive muscle contraction in patients with Lambert-Eaton syndrome.7,8
The distribution of weakness elicited in the history should be confirmed on physical examination with careful attention to proximal vs. distal strength and symmetric vs. asymmetric strength.
EXTRANEUROLOGIC FINDINGS
Extraneurologic findings may help narrow the diagnosis in patients with muscle weakness. As noted earlier, fever suggests inflammatory or infectious myopathy. Cushingoid appearance (buffalo hump, purple striae) suggests endogenous or exogenous glucocorticoid excess. A violaceous rash on the knuckles and extensor surfaces of the upper extremities (Gottron papules; Figure 246 ), a heliotrope rash, the V-sign (violaceous rash on the upper chest and neck), and the shawl sign (violaceous rash on the back) occur with dermatomyositis. Cardiac arrhythmias may occur with inflammatory myopathy, genetic disorders, and electrolyte imbalances.3,9,10,14,26
FIGURE 2.
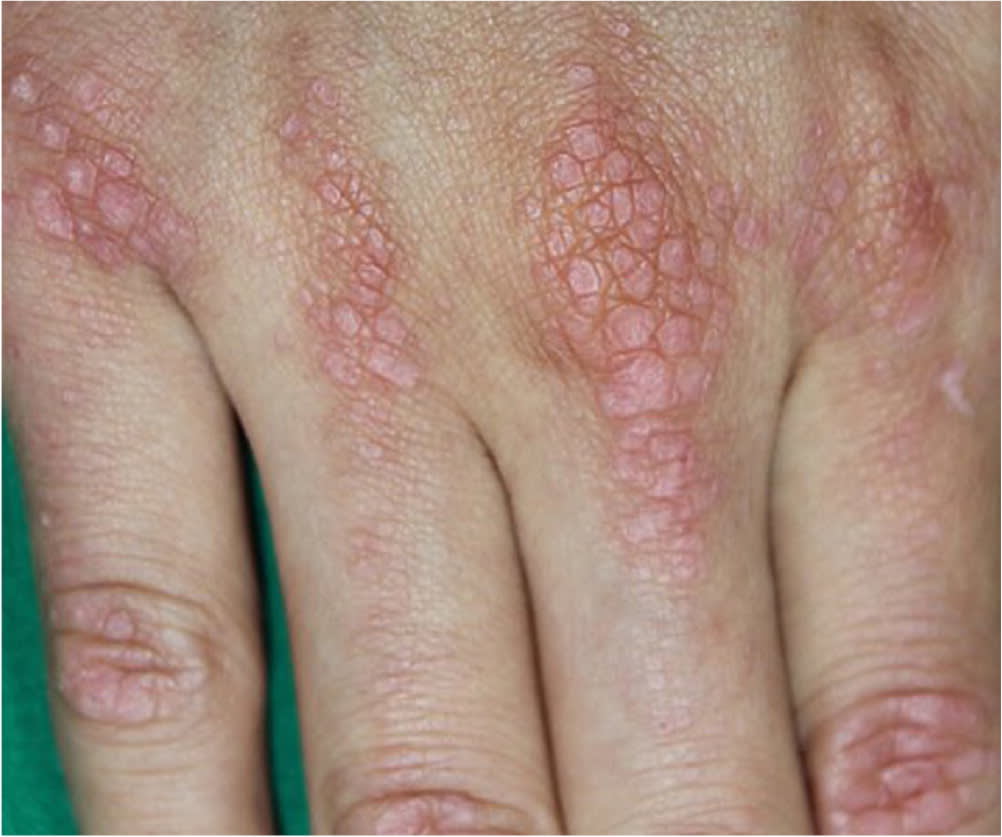
Gottron papules.
Reprinted with permission from Kim HS, Kim SM, Lee JD. Erythematous papules on dorsum of both hands. Am Fam Physician. 2017;95(12):803.
SARCOPENIA AND FRAILTY
Numerous screening tools are available to evaluate weakness in older patients if sarcopenia or frailty is suspected. They include the Fried frailty index, FRAIL (fatigue, resistance, ambulation, illnesses, loss of weight) scale, grip strength analysis, and gait speed analysis.47 Grip strength can be measured objectively with a dynamometer, and gait speed can be measured by timing a patient's usual walking gait to the examination room or by using the Timed Up and Go test.48 Age-specific normative values for grip strength and gait speed are available.38,39
Laboratory Evaluation
Depending on clinical findings and suspected diagnosis, focused or expansive laboratory testing may be indicated in patients with muscle weakness.
Creatine kinase elevation is nonspecific but can help narrow the differential diagnosis and confirm myopathy.26 Creatine kinase levels should not be checked immediately after electrodiagnostic testing because they may be transiently elevated.5,18 Myoglobinuria may also indicate myopathy.3
Because metabolic abnormalities may contribute to weakness, an electrolyte panel should be considered to determine levels of calcium, potassium, sodium, phosphate, and magnesium. Thyroid function testing and cortisol measurement should also be considered.
Serum testing for infectious etiologies, including HIV infection, Lyme disease, West Nile virus infection, and dengue fever, may be indicated if a pathogen is suspected. Lumbar puncture may be indicated if central nervous system infection is suspected.
Anti–acetylcholine receptor antibodies are present in 80% to 90% of patients with generalized myasthenia gravis but in only 50% to 55% of patients with isolated ocular myasthenia gravis.7,11 If clinical suspicion is high despite negative findings on serum anti–acetylcholine receptor antibody testing, the patient should be referred to a neurologist.4,7,11
Specific serum molecular genetic testing may be performed if there is concern for genetic disorders, although this testing is typically ordered by a specialist (e.g., neurologist, rheumatologist, geneticist).
Imaging and Electrodiagnostic Testing
Timely neuroimaging is critical in the evaluation of suspected stroke and cauda equina syndrome. If other neurologic diagnoses (e.g., multiple sclerosis, spinal stenosis, stroke) are suspected, appropriate imaging should be performed. Magnetic resonance imaging and ultrasonography of muscle tissue may be used in the diagnosis of inflammatory myopathy.5
Electromyography should be performed in patients with muscle weakness if the diagnosis is unclear after a history, physical examination, and targeted laboratory evaluation.5,18,19,26 Electromyography assists in localizing the cause of weakness to the motor neuron, neuromuscular junction, or muscle.3 Although electromyography complements the neurologic examination, the findings are not pathognomonic for specific diseases. Abnormal findings on electromyography, with or without muscle imaging, may help localize a suitable site for muscle biopsy.5,18,19
Muscle Biopsy
Muscle biopsy is considered the definitive test for diagnosing myopathies; however, it is invasive and used sparingly. Muscle biopsy should be considered to clarify the diagnosis in patients who have objective weakness with elevated creatine kinase levels or abnormal findings on electromyography or on muscle magnetic resonance imaging.20,21 Biopsy may also be necessary in cases of suspected inflammatory myopathy if the skin findings of dermatomyositis are absent.9,10
Muscle biopsy is generally performed as an outpatient procedure by a surgical consultant. The ideal biopsy site is a muscle that exhibits mild-to-moderate clinical weakness. Severely weak muscles should be avoided, because biopsy of a muscle in an advanced stage of myopathy often shows signs of fibrosis or fatty infiltration, yielding nondiagnostic results.5,9 Muscle biopsy in older patients with significant sarcopenia is unlikely to be diagnostic.5 Complications of muscle biopsy are uncommon but include pain, stiffness, bleeding, and infection.5,21
Data Sources: We searched PubMed, the Cochrane database, and Essential Evidence Plus for the terms muscle weakness, muscle weakness evaluation, myopathy, motor neuropathy, muscle biopsy, neurologic weakness, electrodiagnostic testing weakness, myasthenia gravis, Lambert Eaton, inflammatory myopathy, sarcopenia, frailty, Guillain Barré, Parsonage Turner, hypocalcemia, Chvostek, Trousseau calcium, and multiple sclerosis. Search dates: January 12 to June 13, 2019, and September 22, 2019.
