Henoch-Schönlein purpura, now called immunoglobulin A (IgA) vasculitis, is a systemic, immune complex–mediated, small-vessel leukocytoclastic vasculitis characterized by nonthrombocytopenic palpable purpura, arthritis, and abdominal pain. It is the most common vasculitis in children but can also occur in adults. Diagnostic testing is required only to exclude other etiologies of purpura, to identify renal involvement, and, if indicated, to determine its extent with biopsy. Imaging or endoscopy may be needed to assess organ complications. IgA vasculitis spontaneously resolves in 94% of children and 89% of adults, making supportive treatment the primary management strategy. However, a subset of patients experience renal involvement that can persist and relapse years later. Additional complications can include gastrointestinal bleeding, orchitis, and central nervous system involvement. Systematic reviews have shown that steroids do not prevent complications and should not be used prophylactically. However, randomized trials have demonstrated success with high-dose steroids, cyclosporine, and mycophenolate in treating glomerulonephritis and other complications. Long-term prognosis depends on the extent of renal involvement. Six months of follow-up is prudent to assess for disease relapse or remission.
Immunoglobulin A (IgA) vasculitis, formerly known as Henoch-Schönlein purpura, is defined as a systemic, immune complex–mediated, small-vessel leukocytoclastic vasculitis characterized by nonthrombocytopenic palpable purpura, abdominal pain, and arthritis. It is the most common vasculitis in children. IgA vasculitis is typically self-limited, but a subset of patients experience a remitting-relapsing course. Glomerulonephritis and gastrointestinal bleeding are the most commonly associated complications.
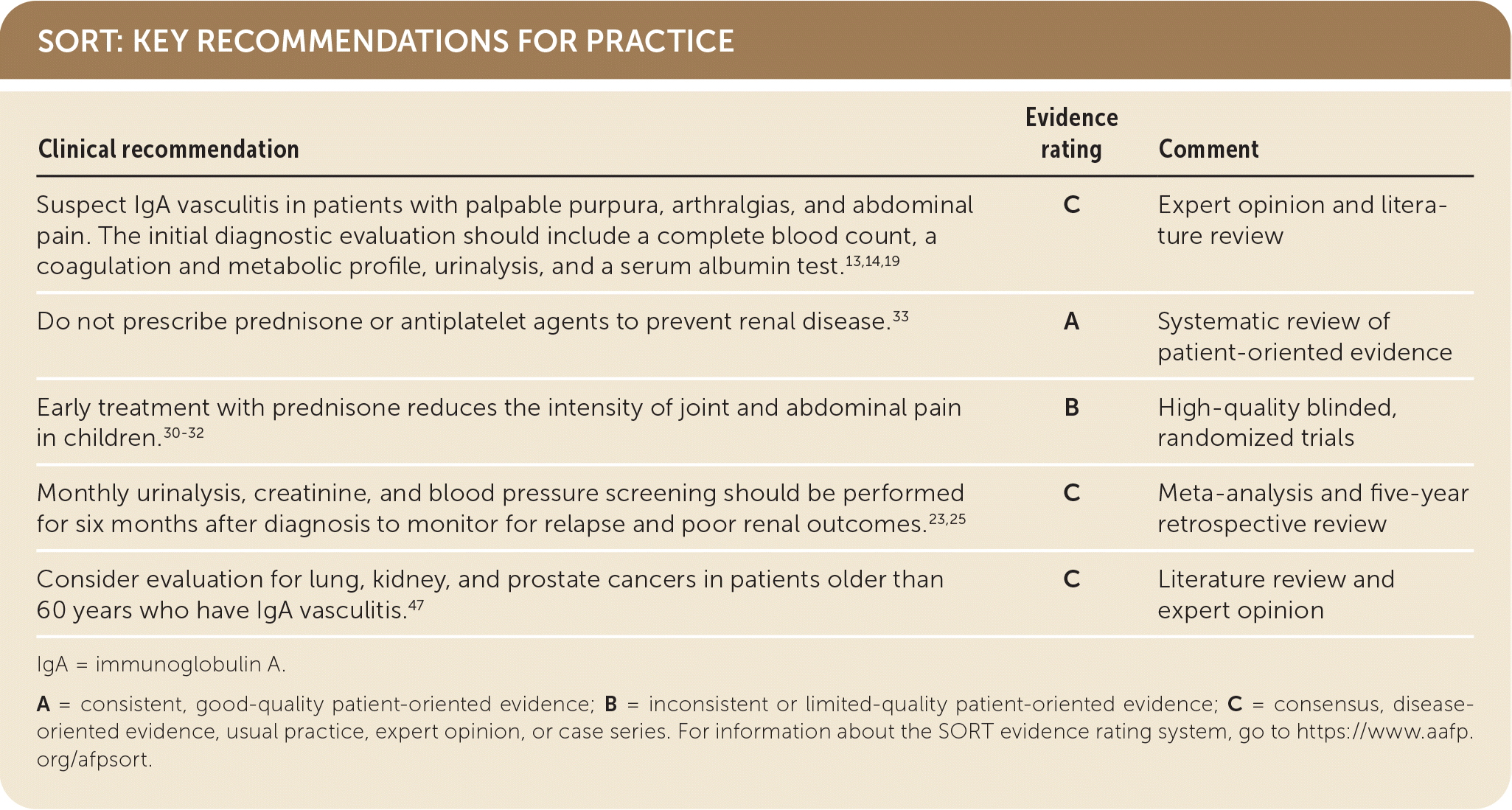
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | Comment |
|---|---|---|
| Suspect IgA vasculitis in patients with palpable purpura, arthralgias, and abdominal pain.The initial diagnostic evaluation should include a complete blood count, a coagulation and metabolic profile, urinalysis, and a serum albumin test.13,14,19 | C | Expert opinion and literature review |
| Do not prescribe prednisone or antiplatelet agents to prevent renal disease.33 | A | Systematic review of patient-oriented evidence |
| Early treatment with prednisone reduces the intensity of joint and abdominal pain in children.30–32 | B | High-quality blinded, randomized trials |
| Monthly urinalysis, creatinine, and blood pressure screening should be performed for six months after diagnosis to monitor for relapse and poor renal outcomes.23,25 | C | Meta-analysis and five-year retrospective review |
| Consider evaluation for lung, kidney, and prostate cancers in patients older than 60 years who have IgA vasculitis.47 | C | Literature review and expert opinion |
IgA = immunoglobulin A.
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.
Epidemiology
- IgA vasculitis occurs in 3.0 to 26.7 out of 100,000 children and 0.8 to 1.8 out of 100,000 adults each year.1
- The mean age of onset in children is six years; onset ranged from 32 to 50 years of age in several adult case series.1–4 More than 90% of cases occur in children younger than 10 years.3
- There is a slight disease predominance in males.1,2
- In children, onset is more common during the fall and winter, but no seasonal pattern has been consistently shown in adults.1,5
- IgA vasculitis is milder in children younger than two years, but more severe in adults, with worse outcomes.3,4,6
Pathophysiology
- IgA vasculitis is a small-vessel vasculitis caused by IgA immune deposits in the gastrointestinal system, joints, skin, and kidneys.
- Multiple viral and bacterial infections are thought to trigger the disease, including Streptococcus, parainfluenza, and human parvovirus B19.7 Cytokines and chemokines are involved; however, the full pathophysiology is not understood.
- Numerous studies have linked disease predisposition, severity, and long-term morbidity with genes on portions of the HLA alleles.8
Diagnosis
- IgA vasculitis should be suspected in patients presenting with palpable purpura who also develop arthralgias (75% of patients) and abdominal pain (50% to 65% of patients).2,9,10
- The differential diagnosis includes immune thrombocytopenic purpura, child abuse, bleeding disorders, medication reactions, senile purpura, meningococcal sepsis, familial Mediterranean fever, Rocky Mountain spotted fever, acute leukemia, bone marrow failure syndromes, and other vasculitides.11,12
- The European League Against Rheumatism/Paediatric Rheumatology European Society diagnostic criteria (Table 113 ) have a 100% sensitivity and 87% specificity in children, and a 99% sensitivity and 86% specificity in adults. These criteria are more accurate than the 1990 American College of Rheumatology criteria.13,14
TABLE 1. Diagnostic Criteria for IgA Vasculitis from the European League Against Rheumatism and the Paediatric Rheumatology European Society

| Mandatory criterion: purpura or petechiae with lower limb predominance |
| Minimum of one out of the four following criteria: Arthritis or arthralgia of acute onset Diffuse abdominal pain with acute onset Histopathology showing leukocytoclastic vasculitis or proliferative glomerulonephritis with IgA deposits Renal involvement (proteinuria or hematuria) |
IgA = immunoglobulin A.
Information from reference 13.
SIGNS AND SYMPTOMS
- Signs and symptoms may develop over days to weeks in any sequence.11 The onset of IgA vasculitis typically follows an upper respiratory infection.15
- Joint involvement and abdominal pain are more common in children, whereas adults are more likely to have lower-extremity edema and hypertension.9
- The characteristic rash may start as erythematous papules that develop into crops of petechiae and palpable purpura.2,9,16 The purpura can enlarge into palpable ecchymoses that evolve from purple to rust-colored as they fade over approximately 10 days.16–18 The purpura are predominantly on extensor surfaces and dependent areas that are subject to pressure, such as the legs and buttocks19 (Figure 1).
- Arthralgias are more common in the knees and ankles than in small joints.11,19 Arthritis is transient and does not damage the joints.17,19
- Abdominal pain is typically colicky and can be severe enough to mimic an acute abdomen.11 Emesis and gastrointestinal bleeding can occur in approximately one-third of patients.17,19 Intussusception can occur in rare cases.20
- Renal disease occurs in 50% of patients and may cause long-term damage.21,22 The risk of renal disease is highest in adults and children older than 10 years.21,23 Males and patients with persisting purpura, abdominal pain, gastrointestinal bleeding, or relapsing episodes are also at increased risk.24 Renal disease typically develops within one to three months after the rash, but it may be delayed up to six months.21,22,24,25 Signs of renal disease include microscopic hematuria, red cell casts, proteinuria, and overt renal failure. Progressive glomerulonephritis may develop, and patients with persistent proteinuria are at highest risk of this complication.19,22
- Low-grade fever and fatigue are common. Less common symptoms include orchitis, pulmonary hemorrhage, and central nervous system involvement with headaches, behavior changes, seizures, or hemorrhage.19,26,27
FIGURE 1.
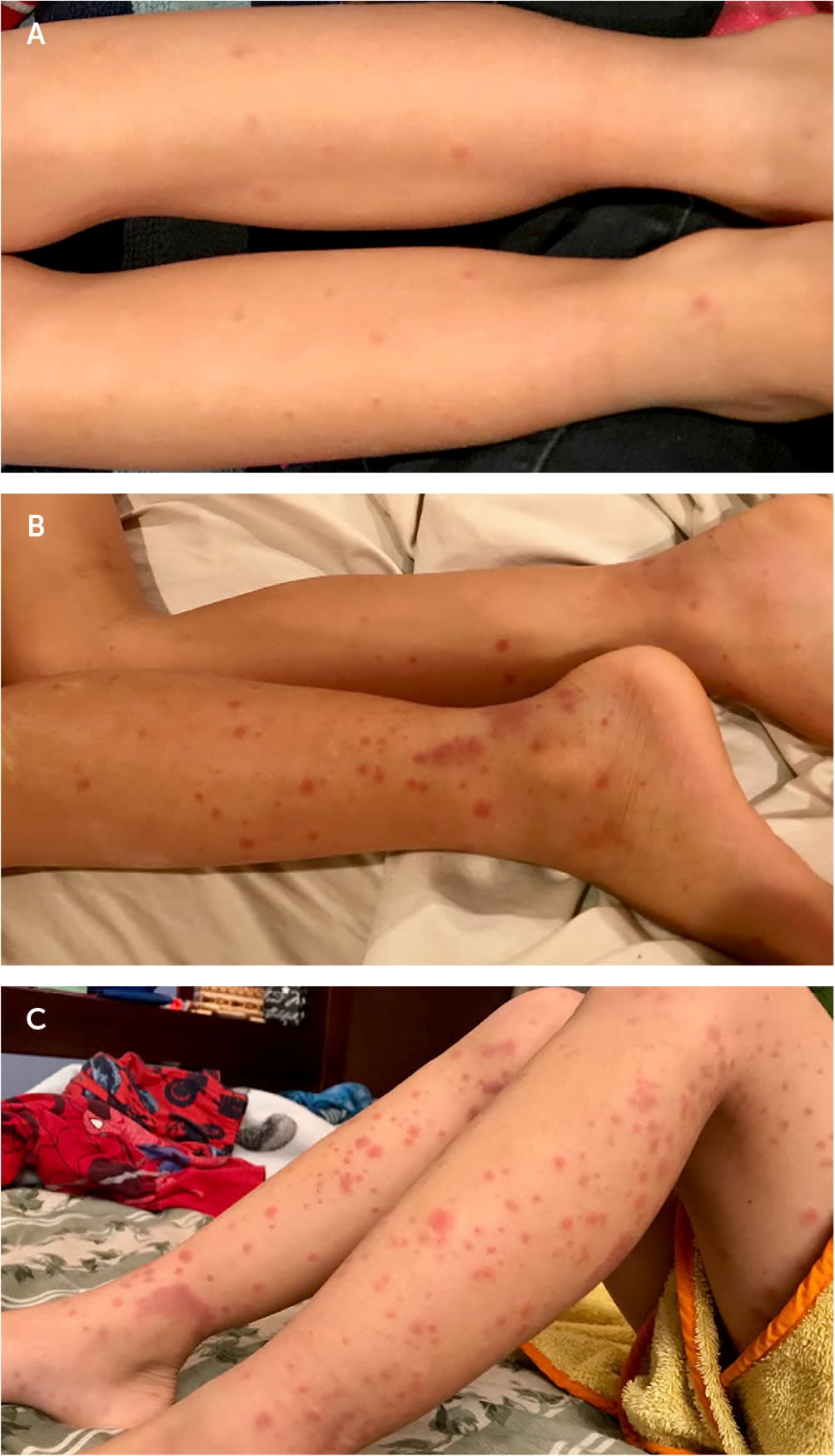
Progression of rash in immunoglobulin A vasculitis. (A) Early stage. (B) Erythematous papules, petechiae, and purpura develop over days to weeks. (C) Petechiae and purpura at different stages of development; note the rust-colored appearance of the older lesions.
DIAGNOSTIC TESTS
- Tests are not required to diagnose IgA vasculitis.
- The laboratory evaluation should exclude other diagnoses and identify disease complications. Useful studies include a complete blood count to exclude blood loss and determine the platelet count; a coagulation profile to exclude coagulopathy; electrolytes and renal function tests to exclude renal disease; and a serum albumin test to assess for intestinal protein loss.17,19 Urinalysis can identify hematuria, casts, or proteinuria. A skin biopsy is needed only in cases where the diagnosis is unclear. IgA vascular deposits are characteristic of the disease.28 A renal biopsy is required only in cases of progressive renal disease.25
- The fecal calprotectin level may be a reliable marker for gastrointestinal involvement.29
- Endoscopy is needed in cases of gastrointestinal or pulmonary hemorrhage. Imaging studies may be required to diagnose the etiology of scrotal enlargement or to evaluate cerebral involvement.
Treatment
- IgA vasculitis spontaneously resolves in 94% of children and 89% of adults, making supportive treatment the primary intervention.11
- The rash requires no specific treatment. Treat arthralgias with oral acetaminophen or nonsteroidal anti-inflammatory drugs (NSAIDs). If there is renal involvement, avoid NSAIDs.
- Renal involvement should prompt nephrology consultation. Although corticosteroids were routinely used based on older studies,30,31 an appropriately powered 2013 double-blind, randomized trial comparing corticosteroids with placebo showed no benefit in reducing proteinuria 12 months after disease onset.32 A 2015 Cochrane review also concluded that steroids and cyclophosphamide do not help prevent renal disease.33
- Randomized trials have shown that joint and abdominal symptoms resolve 1.2 days faster (95% confidence interval, 1.17 to 1.91) in children who receive prednisone, 1 to 2 mg per kg.30–32 Because abdominal symptoms resolve spontaneously, use steroids only in patients with severe pain who do not improve with supportive care and NSAIDs.
- Immunosuppressive therapy (e.g., high-dose intravenous steroids) is often used for the treatment of glomerulonephritis with severe renal involvement.34,35
- Small randomized trials of cyclosporine (Sandimmune) and mycophenolate (Cellcept) have shown success in the treatment of steroid-resistant renal disease.36,37 Dapsone and rituximab (Rituxan) have also shown early success in patients with severe skin and renal involvement.38,39
- Factors predicting the need for hospitalization include orchitis, moderate or severe abdominal pain, arthritis in two or more joints, proteinuria, gastrointestinal bleeding, and inability to ambulate.40
Prognosis
- Relapses most commonly involve the skin but can also involve the joints, kidneys, and gastrointestinal system; occur in 2% to 30% of children; and may occur up to 10 years later.17,41–44
- Gastrointestinal symptoms at diagnosis are the best predictor of relapse in adults.43
- Abnormal urinalysis in children on the day of diagnosis is predictive of severe renal involvement later.45
- No renal involvement in the first six months predicts a low likelihood of chronic disease; 91% of renal involvement occurs within six weeks and 97% within six months.25 Only 2% of children develop nephritis after two months.17
- Nephritis at disease onset raises the risk of hypertension or urine abnormalities for up to eight years, and the risk of proteinuria for five years in children.17,23 Nephrotic syndrome at diagnosis that lasts more than three months carries a risk of long-term renal involvement in children.46
- Any baseline renal disease before IgA nephritis increases the risk of progression to end-stage renal disease; however, less is known about the predictors of renal progression in adults. Adults have an 11% risk of end-stage renal disease and a 13% risk of severe renal failure.35
Follow-up
- Order urinalysis and measure creatinine and blood pressure at least monthly for patients with renal abnormalities at the time of diagnosis.25 The optimal frequency and length of follow-up is unclear; however, a six-month monitoring period is prudent.23,25
- Evaluation for lung, kidney, and prostate cancers should be considered in patients older than 60 years who have IgA vasculitis.47
This article updates previous articles on this topic by Reamy, et al.,11 and by Kraft, et al.48
Data Sources: PubMed was searched using the key term Henoch-Schönlein purpura for human studies and systematic reviews published after January 1, 2009. Reference lists from key articles were reviewed for additional sources. An evidence summary from Essential Evidence Plus was also reviewed. Search dates: May to June 2019, and March 2020.
Figure 1 photos courtesy of Kenny Lin, MD, MPH, Georgetown University, Washington, D.C.
The views expressed in this article are those of the authors and do not reflect the official policy or position of the Department of the Army, Department of the Navy, Department of the Air Force, Uniformed Services University of the Health Sciences, Department of Defense, or the U.S. government.
