Gout is a disease resulting from the deposition of urate crystals caused by the overproduction or underexcretion of uric acid. The disease is often, but not always, associated with elevated serum uric acid levels. Clinical manifestations include acute and chronic arthritis, tophi, interstitial renal disease and uric acid nephrolithiasis. The diagnosis is based on the identification of uric acid crystals in joints, tissues or body fluids. Treatment goals include termination of the acute attack, prevention of recurrent attacks and prevention of complications associated with the deposition of urate crystals in tissues. Pharmacologic management remains the mainstay of treatment. Acute attacks may be terminated with the use of nonsteroidal anti-inflammatory agents, colchicine or intra-articular injections of corticosteroids. Probenecid, sulfinpyrazone and allopurinol can be used to prevent recurrent attacks. Obesity, alcohol intake and certain foods and medications can contribute to hyperuricemia. These potentially exacerbating factors should be identified and modified.
Gout is a disease resulting from the deposition of monosodium urate crystals in synovial fluid and other tissues or the formation of uric acid stones in the kidney. Although the prevalence of gout is equal in men and women, men are six times more likely to have serum uric acid concentrations above 7 mg per dL (420 μmol per L). Gout typically occurs during middle age and is uncommon before the age of 30 years. Women rarely have gouty arthritis attacks before menopause.1
Pathogenesis
Hyperuricemia is defined as a serum uric acid concentration above 7 mg per dL (420 μmol per L). This concentration is also the limit of solubility for monosodium urate in plasma. At levels of 8 mg per dL (480 μmol per L) or greater, monosodium urate is more likely to precipitate in tissues. At a pH of 7, more than 90 percent of uric acid exists as monosodium urate.
Uric acid, the end product of purine metabolism, is a waste product that has no physiologic role. Humans lack uricase, an enzyme that breaks down uric acid into a more water-soluble product (allantoin), thus preventing uric acid accumulation. Increased serum uric acid concentration is a result of either overproduction or underexcretion of uric acid. In 90 percent of patients, gout is caused by the underexcretion of uric acid.2
Although hyperuricemia is a risk factor for the development of gout, the exact relationship between hyperuricemia and acute gout is unclear. Acute gouty arthritis can occur in the presence of normal serum uric acid concentrations. Conversely, many persons with hyperuricemia never experience an attack of gouty arthritis.3
Hyperuricemia can have many causes. Serum uric acid levels become elevated in any disorder that results in the proliferation of cells or the excessive turnover of nucleoproteins. Hyperuricemia can also occur with decreased renal function and in genetic disorders that increase the production or limit the excretion of uric acid (Table 1).4 Several medications increase the serum uric acid concentration through modification of the filtered load of uric acid or one of the tubular transport processes.
Hyperuricemia has been associated with hypertriglyceridemia and diabetes mellitus,5 and it may be a risk factor for the development of coronary artery disease.6 Gout and rheumatoid arthritis do not appear to be associated.7,8
OVERPRODUCTION OF URIC ACID
Purines, which are later metabolized to uric acid, enter a common metabolic pathway by which either nucleic acid or uric acid is produced. Normal production of uric acid is considered to be 600 mg per day in men with normal renal function on a purine-free diet.4 Overproduction of uric acid may occur because of an abnormality in the enzymes that regulate purine metabolism. Two such abnormalities have been documented. An increase in the activity of phosphoribosylpyrophosphate synthetase results in increased uric acid synthesis. A deficiency of hypoxanthine-guanine phosphoribosyltransferase also increases serum uric acid levels.9
A practical approach is to obtain a 24-hour uric acid determination without dietary restriction. A patient on a regular diet who excretes more than 800 mg of uric acid per 24 hours is considered an overproducer.4
UNDEREXCRETION OF URIC ACID
About two thirds to three fourths of all uric acid produced daily is excreted by the kidneys. The gastrointestinal tract eliminates the other one third to one fourth. Under normal conditions, uric acid is filtered in the glomeruli of the kidney, reabsorbed in the proximal tubule and secreted distally. Tubular secretion is almost entirely responsible for the excretion of uric acid. Renal management of uric acid is defective in approximately 98 percent of patients with primary hyperuricemia and gout.4
Clinical Presentation
Initial gout attacks are usually monoarthric. However, polyarthric attacks can also occur. More than 75 percent of acute gout attacks affect a joint in the lower extremity, especially the first metatarsophalangeal joint. Podagra, an acute attack of gout in the great toe, accounts for over 50 percent of all acute attacks (Figure 1). Approximately 85 to 90 percent of patients with gout experience podagra at some point in the disease.10
Patients with recurrent attacks of gout have a longer duration of illness and are more likely to have polyarthric disease. Joint involvement in polyarthric attacks appears to have an ascending, asymmetric pattern. In addition to the great toe, other areas affected include the insteps, heels, ankles, knees, fingers, wrists and elbows.
FIGURE 1.
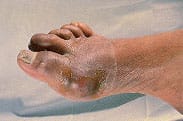
Podagra: monarticular arthritis of the first metatarsophalangeal joint.
ACUTE GOUTY ARTHRITIS
Acute gouty arthritis attacks may occur without provocation, or they may be precipitated by a number of conditions that raise uric acid levels (Table 1).4 Modifiable risk factors for gout attacks include alcohol consumption, obesity, hypertension and occupational and environmental exposure to lead.11
Any abrupt change in the serum uric acid concentration may provoke an acute attack of gouty arthritis. Fluctuations in the serum acid concentration may occur in persons who are fasting, who consume binge amounts of alcohol or who ingest large amounts of protein and purine-rich foods (e.g., bacon, salmon, sweetbreads, scallops, turkey).
The chief complaint associated with an acute attack of gout is agonizing pain accompanied by signs of inflammation, including swelling, erythema, warmth and tenderness. A low-grade fever may occur in conjunction with the inflammation. Acute attacks usually peak within one to two days of symptom onset. Untreated attacks may last seven to 10 days.
Attacks usually start during the night, and moderate pain in a joint is first noticed. The pain becomes persistently worse and has a continuous, gnawing quality. The joints in the great toe and other parts of the lower extremity are generally the first articulations to be affected. These joints are common sites of attack because of lower body temperature and decreased monosodium urate solubility. Lower extremity trauma can also lead to an attack. Trauma induced in weight-bearing joints as a result of routine activities causes synovial effusions during daytime hours. At night, water is reabsorbed from the joint spaces, leaving a supersaturated concentration of monosodium urate.
Pain and inflammation are produced when uric acid crystals activate the humoral and cellular inflammatory processes.12 Because an acute attack begins suddenly, the swelling, erythema and tenderness in a joint may be misdiagnosed as septic arthritis or cellulitis.13
INTERVAL GOUT
Interval or intercritical gout is the condition that occurs after the acute attack has resolved and the patient has become asymptomatic. At this point, the physician usually decides whether or not to initiate prophylactic hyperuricemic therapy. Generally, patients with hyperuricemia and recurrent attacks, chronic gout, tophi, gouty arthritis or nephrolithiasis should be treated. Some investigators argue that the first attack of acute gouty arthritis is grounds for the initiation of hyperuricemic treatment. Others contend that a first attack is easily treated and recommend withholding prophylactic therapy until additional attacks occur.14,15
TOPHACEOUS GOUT
Tophi are nodular masses of monosodium urate crystals deposited in the soft tissues of the body. They are a late complication of hyperuricemia. Rarely, tophi can develop without previous acute gouty arthritis.16 The most common sites of urate deposition are the base of the great toe, and the fingers, wrist, hand, olecranon bursae and Achilles tendon (Figures 2 and 3). Tophi occur, on average, approximately 12 years after the initial attack (reported range: three to 42 years after initial symptoms).2 Complications of tophi include pain, soft tissue damage and deformity, joint destruction and nerve compression syndromes such as carpal tunnel syndrome.
FIGURE 2.
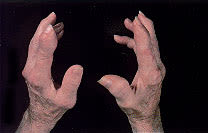
Chronic tophaceous gout, with subcutaneous tophi causing bony deformity of the hands of the patient in Figure 1.
FIGURE 3.
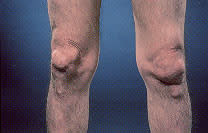
Chronic tophaceous gout, with subcutaneous tophi causing bony deformity of the knees.
RENAL MANIFESTATIONS
The three renal complications of gout are nephrolithiasis and acute and chronic gouty nephropathy. Nephrolithiasis occurs in approximately 10 to 25 percent of patients with primary gout.17 The solubility of uric acid crystals increases as the urine pH becomes more alkaline. Acidic urine saturated with uric acid crystals may result in spontaneous stone formation. Other types of stones may also develop, because uric acid can act as a nidus for calcium oxalate or phosphate stones.
Acute gouty nephropathy usually results from the massive malignant cell turnover that occurs with the treatment of myeloproliferative or lymphoproliferative disorders. The blockage of urine flow secondary to the precipitation of uric acid in the collecting ducts and ureters can lead to acute renal failure.
Long-term deposition of crystals in the renal parenchyma can cause chronic urate nephropathy. The formation of microtophi causes a giant cell inflammatory reaction. This results in proteinuria and inability of the kidney to concentrate urine.2
Diagnostic Evaluation
Because patients with gout typically have hypertension and impaired renal function, examination of the renal and cardiovascular systems is essential. Baseline laboratory tests should include a complete blood cell count, urinalysis, and serum creatinine, blood urea nitrogen and serum uric acid measurements.
Radiography is not very useful in diagnosing initial attacks of acute gouty arthritis. The radiographic findings are generally nonspecific, consisting of soft tissue swelling around a joint. Bony abnormalities indicate the presence of chronic gout. In general, gout must be untreated or inadequately treated for approximately 12 years before chronic arthritis and bony erosions are seen on radiographs. Classic radiologic features of gout include tophi, an overhanging edge of cortex and a “punched-out” erosion of bone with sclerotic borders18 (Figure 4). Mineralization is normal, and joint spaces are preserved. Distribution includes the feet, ankles, knees, hands and elbows.
FIGURE 4.
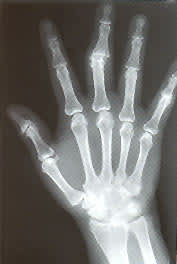
Radiograph showing sharply “punched-out” bony defects of the distal radius and proximal phalynx of the middle finger.
Magnetic resonance imaging (MRI) is not used routinely in the evaluation of gout. Rarely, a gouty tophus may mimic an infectious or neoplastic process. In this instance, MRI evaluation is necessary. Tophaceous gout should be considered when a mass reveals heterogeneously low to intermediate signal intensity, particularly if adjacent bone shows erosive changes or other joints are involved.19
The diagnosis of gout is confirmed by the presence of polymorphonuclear leukocytes and intracellular monosodium urate crystals in synovial fluid aspirated from an inflamed joint (Figure 5). Monosodium urate crystals observed using polarized light microscopy are needle-shaped and negatively birefringent (Figure 6). Examination of aspirated joint fluid can also rule out other disorders that mimic gout, such as septic arthritis and pseudogout. Occasionally, patients with gout may present without uric acid crystals in the synovial fluid aspirate. However, aspiration repeated five hours to one day later shows crystals in the synovial fluid of most of these patients.20,21
FIGURE 5.

Monosodium urate crystals from a tophus, observed with polarized light microscopy.
FIGURE 6.

Intracellular monosodium urate crystal from synovial fluid, observed with polarized light microscopy.
Treatment
Serum uric acid concentrations may be reduced with nonpharmacologic therapy. Useful dietary and lifestyle changes include weight reduction, decreased alcohol ingestion, decreased consumption of foods with a high purine content, and control of hyperlipidemia and hypertension. Used alone, however, these measures will probably not reduce serum uric acid levels to normal, which is the treatment goal for the prevention of acute gout attacks. Symptomatic hyperuricemia usually requires medication.
ACUTE GOUTY ARTHRITIS
Three treatments currently available for acute gouty arthritis attacks are nonsteroidal anti-inflammatory drugs (NSAIDs), colchicine and corticosteroids.
NSAIDs. These rapid-acting drugs are currently the most favored treatment for acute gout attacks. Indomethacin (Indocin) is generally the drug of choice, but other NSAIDs can be used (Table 2). One study reported the achievement of pain relief in patients who presented to an emergency department with acute gouty arthritis who were treated with a 60-mg intramuscular injection of ketorolac (Toradol).22
TABLE 2 Treatment of Gout with Selected Nonsteroidal Anti-inflammatory Drugs*
| Medication | Dosage | Cost per day† |
|---|---|---|
| Indomethacin (Indocin) | 50 mg three times daily | $ 2.75 (0.50 to 1.00) |
| Naproxen sodium (Anaprox) | 825 mg once, then 275 mg every 8 hours | 4.00 (3.50) for first day; 2.50 (2.00 to 2.50 thereafter) |
| Sulindac (Clinoril) | 200 mg twice daily | 2.50 (2.00) |
*—Note that other agents can be used.
†—Estimated cost to the pharmacist based on average wholesale prices (rounded to the nearest quarter dollar) in Red book. Montvale, N.J.: Medical Economics Data, 1999. Cost to the patient will be greater, depending on prescription filling fee.
All NSAIDs can have serious gastrointestinal side effects, including bleeding and ulceration. These drugs should therefore be used with caution in patients with a history of peptic ulcer disease, congestive heart failure or chronic renal failure. Discretion should be used in giving NSAIDs to patients who are allergic to aspirin or have asthma or nasal polyps.
Colchicine. This agent is an effective alternative to NSAIDs in the treatment of acute gouty arthritis. Colchicine is most beneficial when it is given in the first 12 to 36 hours of an attack. It apparently exerts its effect by inhibiting the phagocytosis of uric acid and blocking the release of chemotactic factor. Colchicine has anti-inflammatory activity but no analgesic activity.
Colchicine can be given orally or parenterally. With oral administration, two 0.5- or 0.6-mg tablets are taken initially. Then one tablet is taken every hour until joint symptoms are relieved, gastrointestinal side effects develop (nausea, vomiting and diarrhea) or a total of 5 to 7 mg has been given. Colchicine can be given intravenously in 1-mg doses (not to exceed 4 mg per day) if the oral route is not available or gastrointestinal side effects have to be avoided. Intravenous administration has been associated with an increased risk of toxic side effects, including bone marrow suppression and renal or hepatic cell damage.
Corticosteroids. Monarthric gout responds well to corticosteroids given by intra-articular injection. Systemic corticosteroids (e.g., prednisone [Deltasone], in a dosage of 20 to 30 mg per day) are used only when NSAIDs and colchicine are not effective or are contraindicated.
PREVENTION OF RECURRENT ATTACKS
Hyperuricemic therapy should be initiated in patients with frequent gout attacks, tophi or urate nephropathy. A low dosage of an NSAID or colchicine is effective in preventing acute gouty attacks. Hyperuricemic drug therapy should not be started until an acute attack of gouty arthritis has ended, because of the risk of increased mobilization of uric acid stores. A reasonable goal is to reduce the serum uric acid concentration to less than 6 mg per dL (360 μmol per L).
Uricosuric Drugs. These agents decrease the serum uric acid level by increasing renal excretion. Probenecid (Benemid) and sulfinpyrazone (Anturane) are used in patients who are considered underexcretors of uric acid. Uricosuric drugs should not be given to patients with a urine output of less than 1 mL per minute, a creatinine clearance of less than 50 mL per minute (0.84 mL per second) or a history of renal calculi. The physiologic decline in renal function that occurs with aging frequently limits the use of uricosuric agents.
Probenecid, in a dosage of 1 to 2 g per day, achieves satisfactory control in 60 to 85 percent of patients.23 It is important to note that the drug also blocks the tubular secretion of other organic acids. This may result in increased plasma concentrations of penicillins, cephalosporins, sulfonamides and indomethacin.
Sulfinpyrazone is a uricosuric agent that is related to phenylbutazone. Because it can act as an antiplatelet drug, it should be used cautiously in patients who are anticoagulated or have bleeding problems. Sulfinpyrazone can also cause gastrointestinal problems. Thus, caution should also be exercised in giving this drug to patients with peptic ulcer disease.
Allopurinol. As a xanthine oxidase inhibitor, allopurinol (Zyloprim) impairs the conversion of hypoxanthine to xanthine and the conversion of xanthine to uric acid. The effect of the drug depends on the dosage. Allopurinol in a dosage of 300 mg per day has been reported to reduce serum urate concentrations to less than 7 mg per dL (420 μmol per L) in 70 percent of patients.24
Allopurinol is the drug of choice in patients with severe tophaceous deposits and in patients with a history of impaired renal function (creatinine clearance of less than 50 mL per minute [0.84 mL per second]), uric acid nephropathy or nephrolithiasis. The drug is also preferred as a pretreatment agent to protect against uric acid nephropathy in patients with lymphoproliferative or myeloproliferative disorders.
The side effects of allopurinol include skin rash (e.g., Stevens-Johnson syndrome and toxic epidermal necrolysis), leukopenia and gastrointestinal disturbances. The initiation of allopurinol therapy can also precipitate an acute gout attack. The dosage of allopurinol should be adjusted in patients with renal impairment.
