Thrombocytopenia is defined as a platelet count of less than 150 × 103 per μL. It is often discovered incidentally when obtaining a complete blood count during an office visit. The etiology usually is not obvious, and additional investigation is required. Patients with platelet counts greater than 50 × 103 per μL rarely have symptoms. A platelet count from 30 to 50 × 103 per μL rarely manifests as purpura. A count from 10 to 30 × 103 per μL may cause bleeding with minimal trauma. A platelet count less than 5 × 103 per μL may cause spontaneous bleeding and constitutes a hematologic emergency. Patients who present with thrombocytopenia as part of a multisystem disorder usually are ill and require urgent evaluation and treatment. These patients most likely have an acute infection, heparin-induced thrombocytopenia, liver disease, thrombotic thrombocytopenic purpura/hemolytic uremic syndrome, disseminated intravascular coagulation, or a hematologic disorder. During pregnancy, preeclampsia and the HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome are associated with thrombocytopenia. Patients with isolated thrombocytopenia commonly have drug-induced thrombocytopenia, immune thrombocytopenic purpura, pseudothrombocytopenia, or if pregnant, gestational thrombocytopenia. A history, physical examination, and laboratory studies can differentiate patients who require immediate intervention from those who can be treated in the outpatient setting. Treatment is based on the etiology and, in some cases, treating the secondary cause results in normalization of platelet counts. Consultation with a hematologist should be considered if patients require hospitalization, if there is evidence of systemic disease, or if thrombocytopenia worsens despite initial treatment.
Family physicians often see patients who present with a low platelet count. Although further inquiry may elicit additional signs and symptoms of systemic disease, the etiology of thrombocytopenia usually is not immediately apparent. A low platelet count may be the only hematologic abnormality. In adults, thrombocytopenia is a platelet count less than 150 × 103 per μL (150 × 109 per L). Cases are considered mild if counts are between 70 and 150 × 103 per μL (70 to 150 × 109 per L) and severe if less than 20 × 103 per μL (20 × 109 per L).1 Patients with a platelet count greater than 50 × 103 per μL (50 × 109 per L) often are asymptomatic. Patients with a count from 30 to 50 × 103 per μL (30 to 50 × 109 per L) rarely present with purpura, although they may have excessive bleeding with trauma. However, counts from 10 to 30 × 103 per μL (10 to 30 × 109 per L) may cause bleeding with minimal trauma, and counts less than 10 × 103 per μL increase the risk of spontaneous bleeding, petechiae, and bruising. Spontaneous bleeding (i.e., mucosal, intracranial, gastrointestinal, and genitourinary bleeding) is more likely in patients with platelet counts less than 5 × 103 per μL (5 × 109 per L), and is considered a hematologic emergency.2
SORT: KEY RECOMMENDATION FOR PRACTICE
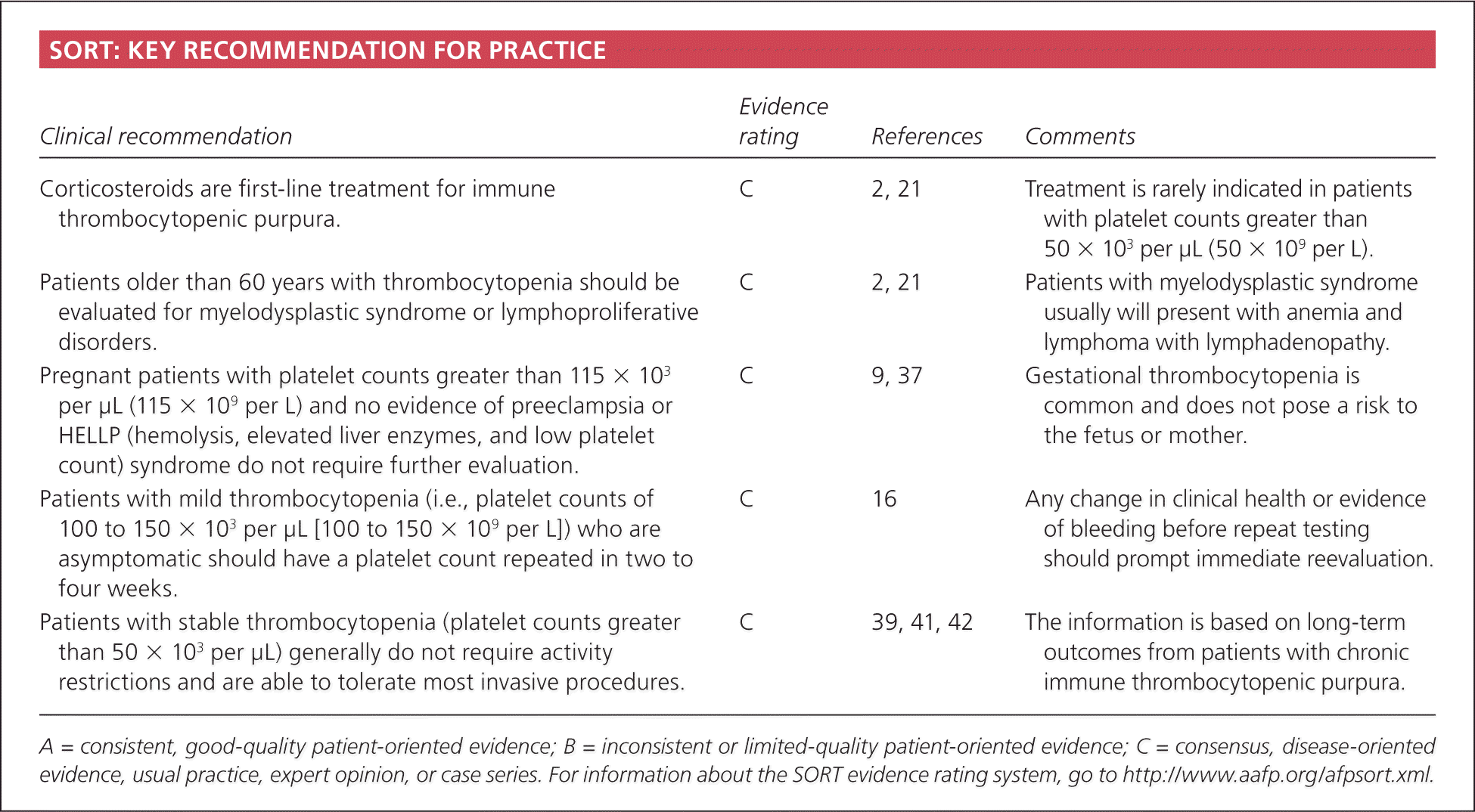
| Clinical recommendation | Evidence rating | References | Comments |
|---|---|---|---|
| Corticosteroids are first-line treatment for immune thrombocytopenic purpura. | C | 2, 21 | Treatment is rarely indicated in patients with platelet counts greater than 50 × 103 per μL (50 × 109 per L). |
| Patients older than 60 years with thrombocytopenia should be evaluated for myelodysplastic syndrome or lymphoproliferative disorders. | C | 2, 21 | Patients with myelodysplastic syndrome usually will present with anemia and lymphoma with lymphadenopathy. |
| Pregnant patients with platelet counts greater than 115 × 103 per μL (115 × 109 per L) and no evidence of preeclampsia or HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome do not require further evaluation. | C | 9, 37 | Gestational thrombocytopenia is common and does not pose a risk to the fetus or mother. |
| Patients with mild thrombocytopenia (i.e., platelet counts of 100 to 150 × 103 per μL [100 to 150 × 109 per L]) who are asymptomatic should have a platelet count repeated in two to four weeks. | C | 16 | Any change in clinical health or evidence of bleeding before repeat testing should prompt immediate reevaluation. |
| Patients with stable thrombocytopenia (platelet counts greater than 50 × 103 per μL) generally do not require activity restrictions and are able to tolerate most invasive procedures. | C | 39, 41, 42 | The information is based on long-term outcomes from patients with chronic immune thrombocytopenic purpura. |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.xml.
Initial Evaluation
Thrombocytopenia can result from decreased platelet production, increased platelet consumption, or sequestration (Table 13–6 ). Common etiologies with clinical findings and suggested treatment are listed in Table 2,7–14 and clinical considerations to aid in diagnosis are listed in Table 3. A systematic approach should be used to evaluate incidental thrombocytopenia. During the patient history, physicians should inquire about easy bruising or petechiae, melena, rashes, fevers, and bleeding. They also should inquire about medication use, immunizations, recent travel, transfusion history, family history, and medical history. A history of acute and chronic alcohol use should be obtained. Any recent hospitalization or heparin exposure should raise the possibility of heparin-induced thrombocytopenia. Pregnant patients with visual symptoms, headaches, abdominal pain, or influenza-like symptoms may have preeclampsia or HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome.
Table 1. Etiologies of Thrombocytopenia

| Decreased platelet production |
| Bone marrow failure (e.g., aplastic anemia, paroxysmal nocturnal hemoglobinuria, Shwachman-Diamond syndrome) |
| Bone marrow suppression (e.g., from medication, chemotherapy, or irradiation) |
| Chronic alcohol abuse* |
| Congenital macrothrombocytopenias (e.g., Alport syndrome, Bernard-Soulier syndrome, Fanconi anemia, platelet-type or pseudo–von Willebrand disease, Wiskott-Aldrich syndrome) |
| Infection† (e.g., cytomegalovirus, Epstein-Barr virus, hepatitis C virus, HIV, mumps, parvovirus B19, rickettsia, rubella, varicella-zoster virus) |
| Myelodysplastic syndrome |
| Neoplastic marrow infiltration |
| Nutritional deficiencies (vitamin B12 and folate) |
| Increased platelet consumption |
| Alloimmune destruction (e.g., posttransfusion, neonatal, posttransplantation) |
| Autoimmune syndromes (e.g., antiphospholipid syndrome, systemic lupus erythematosus, sarcoidosis) |
| Disseminated intravascular coagulation*/severe sepsis* |
| Drug-induced thrombocytopenia |
| Heparin-induced thrombocytopenia |
| Immune thrombocytopenic purpura* |
| Infection † (e.g., cytomegalovirus, Epstein-Barr virus, hepatitis C virus, HIV, mumps, parvovirus B19, rickettsia, rubella, varicella-zoster virus) |
| Mechanical destruction (e.g., aortic valve, mechanical valve, extracorporeal bypass) |
| Preeclampsia/HELLP syndrome |
| Thrombotic thrombocytopenic purpura/hemolytic uremic syndrome |
| Sequestration/other |
| Chronic alcohol abuse* |
| Dilutional thrombocytopenia (e.g., hemorrhage, excessive crystalloid infusion) |
| Gestational thrombocytopenia |
| Hypersplenism (e.g., distributional thrombocytopenia) |
| Liver disease (e.g., cirrhosis, fibrosis, portal hypertension) |
| Pseudothrombocytopenia |
| Pulmonary emboli |
| Pulmonary hypertension |
HELLP = hemolysis, elevated liver enzymes, and low platelet count; HIV = human immunodeficiency virus.
*—More than one mechanism of action.
†—Thrombocytopenia with infection is usually caused by bone marrow suppression. In some cases, the thrombocytopenia is also immune-mediated.
Table 2. Common Etiologies of Thrombocytopenia with Clinical Findings and Suggested Treatment
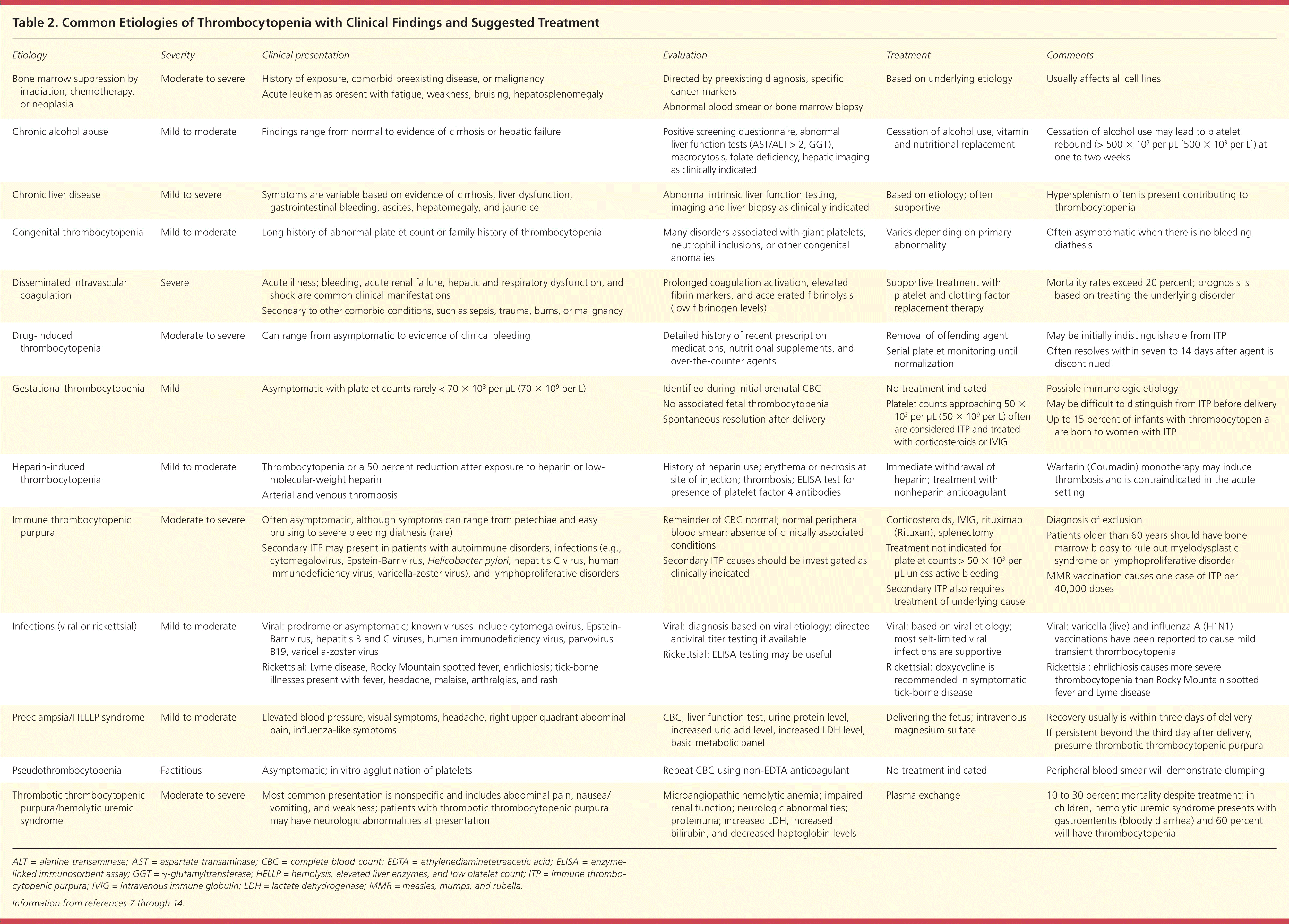
| Etiology | Severity | Clinical presentation | Evaluation | Treatment | Comments |
|---|---|---|---|---|---|
| Bone marrow suppression by irradiation, chemotherapy, or neoplasia | Moderate to severe |
|
|
|
|
| Chronic alcohol abuse | Mild to moderate |
|
|
|
|
| Chronic liver disease | Mild to severe |
|
|
|
|
| Congenital thrombocytopenia | Mild to moderate |
|
|
|
|
| Disseminated intravascular coagulation | Severe |
|
|
|
|
| Drug-induced thrombocytopenia | Moderate to severe |
|
|
|
|
| Gestational thrombocytopenia | Mild |
|
|
|
|
| Heparin-induced thrombocytopenia | Mild to moderate |
|
|
|
|
| Immune thrombocytopenic purpura | Moderate to severe |
|
|
|
|
| Infections (viral or rickettsial) | Mild to moderate |
|
|
|
|
| Preeclampsia/HELLP syndrome | Mild to moderate |
|
|
|
|
| Pseudothrombocytopenia | Factitious |
|
|
|
|
| Thrombotic thrombocytopenic purpura/hemolytic uremic syndrome | Moderate to severe |
|
|
|
|
ALT = alanine transaminase; AST = aspartate transaminase; CBC = complete blood count; EDTA = ethylenediaminetetraacetic acid; ELISA = enzyme-linked immunosorbent assay; GGT = γ-glutamyltransferase; HELLP = hemolysis, elevated liver enzymes, and low platelet count; ITP = immune thrombocytopenic purpura; IVIG = intravenous immune globulin; LDH = lactate dehydrogenase; MMR = measles, mumps, and rubella.
Table 3. Clinical Considerations to Aid in Diagnosis of Thrombocytopenia

| Clinical consideration | Possible diagnosis |
|---|---|
| Timing | |
| Acute | Acute infection (primarily viral), acute leukemias, aplastic anemia, chemotherapy or irradiation, drug-induced thrombocytopenia, HELLP syndrome/preeclampsia, heparin-induced thrombocytopenia, ITP, malignancies with marrow infiltration, myelofibrosis, TTP/HUS |
| Chronic | Chronic alcohol abuse, congenital syndromes, ITP, liver disease, myelodysplastic syndrome |
| History | |
| Family history | Congenital thrombocytopenia |
| Liver disease | Chronic alcohol abuse, chronic liver disease |
| Pregnancy | Gestational thrombocytopenia, HELLP syndrome/preeclampsia |
| Recent change in medication | Drug-induced thrombocytopenia |
| Recent hospitalization | Heparin-induced thrombocytopenia |
| Recent immunization | MMR, varicella, influenza A (H1N1) |
| Recent transfusion or high-risk behavior | Alloimmune destruction, posttransfusion purpura, viral infection (HCV, HIV) |
| Recent travel | Dengue fever, malaria, rickettsial diseases |
| Recent valve replacement surgery | Mechanical destruction |
| Symptoms | |
| Abdominal pain | HELLP syndrome/preeclampsia, HUS, platelet sequestration |
| Recent fever | Viral infections (CMV, EBV, HIV, influenza A [H1N1], parvovirus B19), TTP, rickettsial diseases |
| Weight loss or night sweats | HIV, malignancies (acute leukemias, myelodysplastic syndrome) |
| Physical examination | |
| Acute rash | Rickettsial diseases, SLE, viral infections |
| Generalized lymphadenopathy | Viral infections (CMV, EBV, HIV), SLE, myeloproliferative disorders, lymphoproliferative disorders |
| Hepatomegaly | Chronic liver disease, acute leukemias, viral infections (CMV, EBV, HBV, HCV) |
| Neurologic | TTP |
| Splenomegaly* | Autoimmune (SLE, sarcoidosis), hypersplenism, viral infections |
CMV = cytomegalovirus; EBV = Epstein-Barr virus; HBV = hepatitis B virus; HCV = hepatitis C virus; HELLP = hemolysis, elevated liver enzymes, and low platelet count; HIV = human immunodeficiency virus; HUS = hemolytic uremic syndrome; ITP = immune thrombocytopenic purpura; MMR = measles, mumps, and rubella; SLE = systemic lupus erythematosus; TTP = thrombotic thrombocytopenic purpura.
*—Ultrasonography may be useful in patients who are obese.
The physical examination should include the eyes (e.g., hemorrhage is suggestive of central nervous system bleeding), abdomen (e.g., splenomegaly, hepatomegaly), lymph nodes (e.g., lymphadenopathy), skin (e.g., petechiae, purpura, bruising), and neurologic system. Bleeding (e.g., epistaxis, mucosal, gastrointestinal, genitourinary) also should be assessed. Thrombocytopenia can be classified as emergent (usually requires inpatient management) or nonemergent (outpatient management). Some syndromes may fall in either category based on the severity of thrombocytopenia.
A peripheral blood smear can provide diagnostic information on a variety of white blood cell disorders, hemolytic anemias, and thrombocytopenia.15 A blood smear should be obtained during the initial evaluation. Table 4 lists common findings on peripheral blood smear and their associated diagnoses.
Table 4. Common Peripheral Blood Smear Findings and Possible Diagnoses
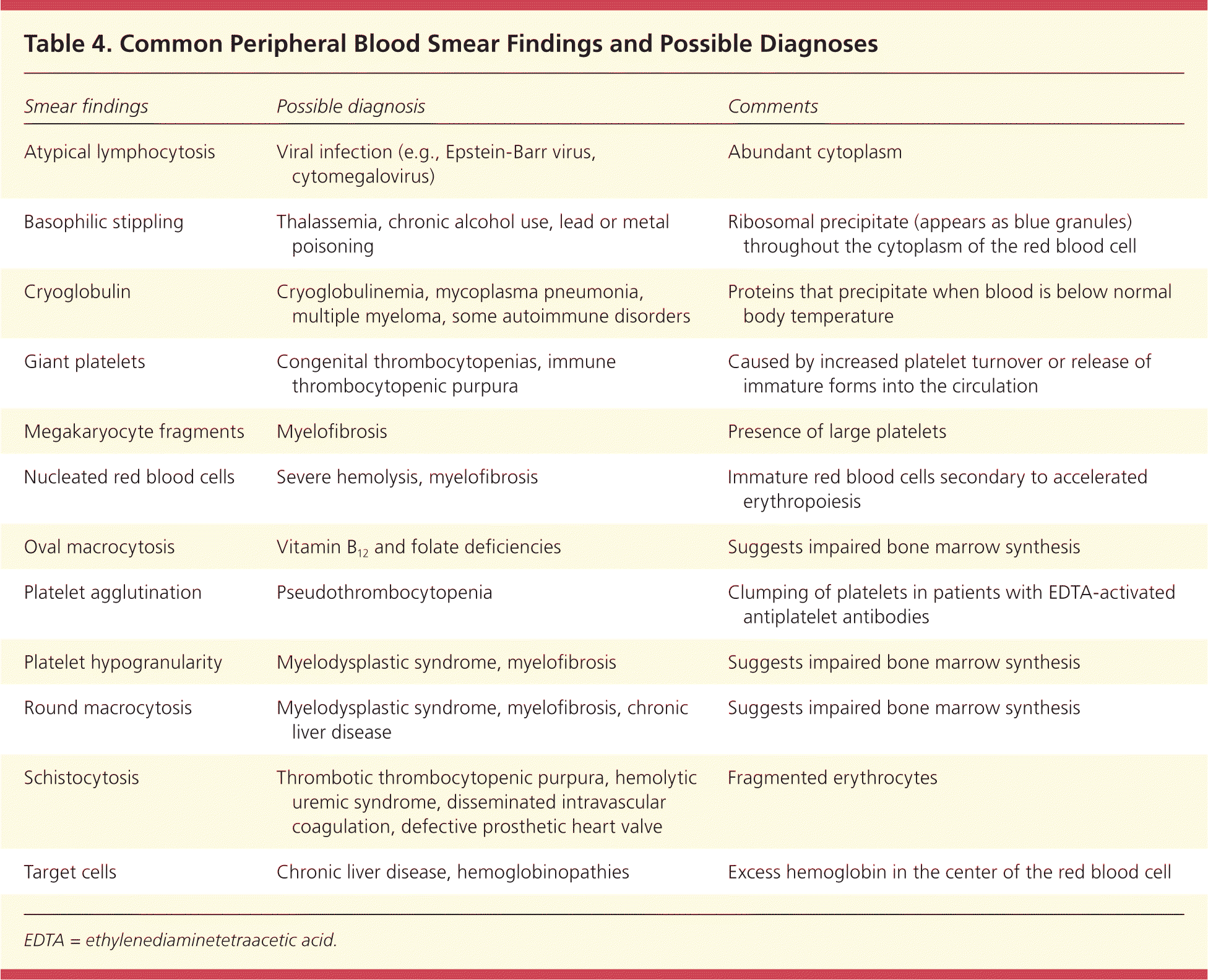
| Smear findings | Possible diagnosis | Comments |
|---|---|---|
| Atypical lymphocytosis | Viral infection (e.g., Epstein-Barr virus, cytomegalovirus) | Abundant cytoplasm |
| Basophilic stippling | Thalassemia, chronic alcohol use, lead or metal poisoning | Ribosomal precipitate (appears as blue granules) throughout the cytoplasm of the red blood cell |
| Cryoglobulin | Cryoglobulinemia, mycoplasma pneumonia, multiple myeloma, some autoimmune disorders | Proteins that precipitate when blood is below normal body temperature |
| Giant platelets | Congenital thrombocytopenias, immune thrombocytopenic purpura | Caused by increased platelet turnover or release of immature forms into the circulation |
| Megakaryocyte fragments | Myelofibrosis | Presence of large platelets |
| Nucleated red blood cells | Severe hemolysis, myelofibrosis | Immature red blood cells secondary to accelerated erythropoiesis |
| Oval macrocytosis | Vitamin B12 and folate deficiencies | Suggests impaired bone marrow synthesis |
| Platelet agglutination | Pseudothrombocytopenia | Clumping of platelets in patients with EDTA-activated antiplatelet antibodies |
| Platelet hypogranularity | Myelodysplastic syndrome, myelofibrosis | Suggests impaired bone marrow synthesis |
| Round macrocytosis | Myelodysplastic syndrome, myelofibrosis, chronic liver disease | Suggests impaired bone marrow synthesis |
| Schistocytosis | Thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, disseminated intravascular coagulation, defective prosthetic heart valve | Fragmented erythrocytes |
| Target cells | Chronic liver disease, hemoglobinopathies | Excess hemoglobin in the center of the red blood cell |
EDTA = ethylenediaminetetraacetic acid.
Long-term Outcomes of Incidental Thrombocytopenia
The findings of isolated thrombocytopenia in an outpatient setting have prognostic implications. A prospective study evaluated the long-term outcomes of patients with incidental thrombocytopenia. The study followed 217 persons with platelet counts from 100 to 150 × 103 per μL (100 to 150 × 109 per L) over a 10-year period. In 64 percent of patients, platelet counts normalized or remained stable. The probability of developing immune thrombocytopenic purpura or an autoimmune disorder was approximately 7 and 12 percent, respectively. Four cases of myelodysplastic syndrome were diagnosed (2 percent), all of which were in older patients.16
Factitious Thrombocytopenia
Pseudothrombocytopenia is secondary to platelet clumping and has no clinical significance. It occurs in one in 1,000 persons in the general population, and can be confirmed by a peripheral blood smear.3 Causes include use of abciximab (Reopro) or ethylenediaminetetraacetic acid–dependent agglutinins.17,18 The platelet count should be repeated by collecting blood in a tube with a non-ethylenediaminetetraacetic acid anticoagulant, such as heparin or sodium citrate. If the complete blood count still shows thrombocytopenia, other causes should be investigated.
Emergent Thrombocytopenia
IMMUNE (IDIOPATHIC) THROMBOCYTOPENIC PURPURA
Immune thrombocytopenic purpura is an acquired immune-mediated disorder characterized by isolated thrombocytopenia and the absence of other conditions or agents known to induce thrombocytopenia. The incidence is 100 cases per 1 million persons annually, and approximately 50 percent of cases occur in children.2 Immune thrombocytopenic purpura in children often resolves spontaneously but tends to be more insidious and chronic in adults. The risk of bleeding correlates to the severity of thrombocytopenia. Patients may present without symptoms, with minimal bleeding, or with serious hemorrhage (e.g., mucosal, intracranial, gastrointestinal, genitourinary). Older patients, patients on antiplatelet therapy, and patients with comorbid conditions may have more severe bleeding manifestations.19
Secondary immune thrombocytopenic purpura is associated with other underlying conditions, such as autoimmune disorders (e.g., systemic lupus erythematosus, antiphospholipid syndrome, Graves disease, sarcoidosis), lymphoproliferative disorders, and infections (e.g., human immunodeficiency virus, Epstein-Barr virus, cytomegalovirus, varicella-zoster virus, hepatitis C virus, Helicobacter pylori). Testing to rule out other causes should be performed as clinically indicated. Forty percent of patients with immune thrombocytopenic purpura test positive for antinuclear or antiphospholipid antibodies without having an underlying autoimmune syndrome.20 Treatment is generally restricted to those with severe thrombocytopenia and is rarely indicated when platelet counts are greater than 50 × 103 per μL unless there is evidence of active bleeding. Corticosteroids are considered first-line treatment and increase platelet counts usually within one week of initiation.2,21 Intravenous immune globulin and rituximab (Rituxan) also have been used for initial treatment of immune thrombocytopenic purpura. Second-line treatment includes thrombopoietin-receptor agonists and splenectomy. Any patient older than 60 years presenting with isolated thrombocytopenia should be evaluated for myelodysplastic syndrome and lympho-proliferative disorders.2,21
HEPARIN-INDUCED THROMBOCYTOPENIA
Heparin-induced thrombocytopenia should be suspected in patients recently treated with heparin. Platelet counts decline within five to 10 days in patients with no previous exposure to heparin and may decline precipitously (within hours) in patients with recent heparin exposure. This life-threatening disorder is characterized by the presence of platelet-activating antibodies recognizing multimolecular complexes bound to unfractionated heparin or low-molecular-weight heparin. Patients with heparin-induced thrombocytopenia present with mild thrombocytopenia or a 50 percent decrease in platelet count from baseline.22 The incidence is higher among surgical patients than medical patients. Thrombosis complications (termed heparin-induced thrombocytopenia with thrombosis) develop in 20 to 50 percent of patients and may affect arterial and venous systems, even after heparin is discontinued.23
Characteristic features are erythematous or necrotizing skin reactions at the site of injection or severe manifestations, such as deep venous thrombosis, pulmonary emboli, stroke, or myocardial infarction. The most widely available diagnostic test is an enzyme-linked immunosorbent assay with platelet factor 4/anion complex as the antigen. This test has a high sensitivity (greater than 97 percent) but a lower specificity (74 to 86 percent) because of the presence of platelet factor 4 antibodies in patients without heparin-induced thrombocytopenia.22,24 If the clinical diagnosis is suspected or confirmed by laboratory analysis, heparin should be stopped immediately and treatment should be changed to a nonheparin anticoagulant. One study showed that patients who developed heparin-induced thrombocytopenia had a 20 percent in-hospital mortality rate, regardless of thrombosis development.25
THROMBOTIC THROMBOCYTOPENIC PURPURA
Patients presenting with thrombocytopenia and microangiopathic hemolytic anemia should be admitted to the hospital with a presumptive diagnosis of thrombotic thrombocytopenic purpura. Renal manifestations, neurologic changes, and fever also may be present. Severity is reflected by the extent of microvascular aggregation of platelets resulting in ischemia and necrosis of tissue cells. The incidence of thrombotic thrombocytopenic purpura is four to 11 patients per 1 million annually in the United States.7 The condition often is caused by the absence or deficiency of a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13.7 It is fatal without treatment; prompt initiation of plasma exchange is critical. Thrombotic thrombocytopenic purpura occurs primarily in adults. Hemolytic uremic syndrome is thrombotic thrombocytopenic purpura found in children presenting with acute renal failure, bloody diarrhea, and/or abdominal pain. Shiga toxin-producing Escherichia coli is the most common causative organism in hemolytic uremic syndrome.26
PREECLAMPSIA AND HELLP SYNDROME
Pregnant patients beyond 20 weeks' gestation with thrombocytopenia or signs and symptoms such as headache, visual disturbances, right upper quadrant abdominal pain, or elevated blood pressure should be evaluated for preeclampsia and HELLP syndrome. Laboratory abnormalities may include anemia, elevated liver enzymes, elevated lactate dehydrogenase, and proteinuria.27 Treatment is intravenous magnesium sulfate and active management to deliver the fetus.28 Pregnant patients also can present with other causes of thrombocytopenia, including disseminated intravascular coagulation (as seen with sepsis and placenta abruptio) and thrombotic thrombocytopenic purpura, which can mimic HELLP syndrome.
OTHER CAUSES
Aplastic anemia, chemotherapy, irradiation, acute leukemias, and myelodysplastic disorders may result in severe or symptomatic thrombocytopenia. Paroxysmal nocturnal hemoglobinuria is associated with hemolysis, renal disease, and thrombosis complications. Hospitalized patients with comorbid conditions, such as sepsis, trauma, burns, or malignancy, can develop disseminated intravascular coagulation with resultant thrombocytopenia.
Nonemergent Thrombocytopenia
DRUG-INDUCED THROMBOCYTOPENIA
One of the most common types of thrombocytopenia in the outpatient setting is drug-induced thrombocytopenia. An epidemiologic study from Europe and the United States showed an annual incidence of 10 cases per 1 million persons, but numbers could be higher in older persons and in hospitalized patients.29 Drug-induced thrombocytopenia should be suspected in all patients presenting with an acute drop in platelet count. Most patients with drug-induced thrombocytopenia have moderate to severe thrombocytopenia reaching a nadir platelet count of 20 × 103 per μL.8 Physicians should ask specific questions about the use of any medications, including nutritional supplements and over-the-counter remedies. Quinine is a common cause of drug-induced thrombocytopenia and often is missed in the patient history.
Table 5 lists common medications that may cause thrombocytopenia.8 A comprehensive list of all case reports describing drug-induced thrombocytopenia is available at http://www.ouhsc.edu/platelets. Drug-induced thrombocytopenia typically occurs within five to seven days of exposure to the causative agent and usually resolves within seven to 14 days after discontinuation.30 If drug-induced thrombocytopenia is suspected, the causative medication should be discontinued and the patient's platelet count should be repeated in one week.
Table 5. Medications/Substances Commonly Associated with Drug-Induced Thrombocytopenia

| Medication/substance | Drug class | Proposed mechanism of action |
|---|---|---|
| Abciximab (Reopro) | Glycoprotein IIb/IIIa inhibitor | Drug-specific antibody |
| Alcohol | Ethanol | Suppression of megakaryocyte production |
| Carbamazepine (Tegretol) | Anticonvulsant | Unknown |
| Cephalosporins | Antibiotic | Hapten-dependent |
| Cimetidine (Tagamet) | Histamine H2 blocker | Unknown |
| Eptifibatide (Integrilin) | Glycoprotein IIb/IIIa inhibitor | Drug-specific antibody |
| Gold salts | Antirheumatic | Autoantibody |
| Heparin | Anticoagulant | Platelet factor 4/heparin antibody |
| Hydrochlorothiazide | Antihypertensive | Suppression of megakaryocyte production |
| Interferon | Antiviral | Drug-specific antibody |
| Measles, mumps, and rubella vaccine | Vaccine | Vaccine-specific antibody |
| Phenytoin (Dilantin) | Anticonvulsant | Unknown |
| Procainamide | Antiarrhythmic | Autoantibody |
| Quinidine/quinine | Cinchona alkaloid | Drug-glycoprotein complex |
| Rifampin | Antibiotic | Autoantibody |
| Sulfasalazine (Azulfidine) | Anti-inflammatory | Drug-specific antibody |
| Trimethoprim/sulfamethoxazole (Bactrim, Septra) | Antibiotic | Drug-glycoprotein complex |
| Vancomycin | Antibiotic | Drug-specific antibody |
Adapted with permission from Kenney B, Stack G. Drug-induced thrombocytopenia. Arch Pathol Lab Med. 2009;133(2):314. Copyright 2009. College of American Pathologists.
INFECTIONS
Infections may cause thrombocytopenia by direct bone marrow suppression or increased peripheral platelet consumption. Common viruses include hepatitis B and C, human immunodeficiency virus, Epstein-Barr, cytomegalovirus, parvovirus B19, varicellazoster, rubella, and mumps. Recent studies found thrombocytopenia in 14 percent of hospitalized patients with influenza A (H1N1) virus.31 Platelet counts usually recover in patients with a self-limited viral infection. If the patient has a recent travel history, especially to South America or Mexico, dengue fever and malaria should be considered. Other travel-associated infections, such as typhoid fever , can cause thrombocytopenia.32 Ehrlichiosis, Rocky Mountain spotted fever, and Lyme disease are common tick-borne illnesses that can cause transient thrombocytopenia.33
LIVER DISEASE
Chronic liver disease usually causes persistent thrombocytopenia, and manifests as cirrhosis, fibrosis, and portal hypertension. The most common cause is chronic alcohol abuse; however, other etiologies include infectious hepatitis, drug-induced liver disease, nonalcoholic liver disease, and metabolic disorders. Patients who consume excessive amounts of alcohol can present with varying degrees of liver impairment ranging from asymptomatic fatty liver to end-stage liver disease. Thrombocytopenia results from direct toxic marrow suppression and splenic sequestration. Folic acid deficiency (related to malnutrition) often coexists with alcohol abuse. Abstinence and nutritional replacement often lead to platelet normalization in three to four weeks in the absence of chronic liver disease.34
GESTATIONAL THROMBOCYTOPENIA
Gestational thrombocytopenia is the most common diagnosis of thrombocytopenia in pregnancy, occurring in 8 percent of pregnancies.9,10 Its etiology is thought to be hemodilution and accelerated platelet clearance. Gestational thrombocytopenia is characterized by mild thrombocytopenia, no history of thrombocytopenia, no association of fetal thrombocytopenia, and spontaneous resolution after delivery.35,36 Gestational thrombocytopenia is a benign clinical condition not associated with maternal or neonatal morbidity or mortality.
Recommendations for the initial evaluation of thrombocytopenia in pregnancy include a complete blood count and peripheral blood smear.10 Additional studies are determined on the basis of history and physical examination. Pregnant patients with platelet counts greater than 115 × 103 per μL (115 × 109 per L) and no evidence of preeclampsia or HELLP syndrome do not require further evaluation.9,37
Mild immune thrombocytopenic purpura and gestational thrombocytopenia often are indistinguishable and cannot be differentiated on the basis of antiplatelet antibody screening; however, 12 to 15 percent of neonates delivered to women with immune thrombocytopenic purpura will develop platelet counts of less than 50 × 103 per μL.38 The platelet count in infants should be monitored when immune thrombocytopenic purpura is suspected in the mother.
Management
A suggested algorithm for the management of thrombocytopenia is shown in Figure 1.4 Symptomatic patients require immediate evaluation. Patients with incidental thrombocytopenia (i.e., platelet counts of 50 to 150 × 103 per μL) who are asymptomatic should have a platelet count repeated in one to four weeks depending on the severity of thrombocytopenia.16 Reassessment should be performed immediately if patients become symptomatic during the surveillance period. Additional laboratory evaluation should be performed as clinically indicated, and a trial of discontinuing agents known to decrease platelet counts is recommended. Patients with chronic alcohol abuse or known nutritional deficiencies with stable thrombocytopenia can be treated by a primary care physician. Rickettsial diseases and most viral infections causing transient thrombocytopenia also can be managed by a primary care physician. Patients with unexplained severe thrombocytopenia, decreasing platelet counts, additional hematologic abnormalities, or associated bleeding complications should be referred to a hematologist.
Figure 1 Management of Thrombocytopenia
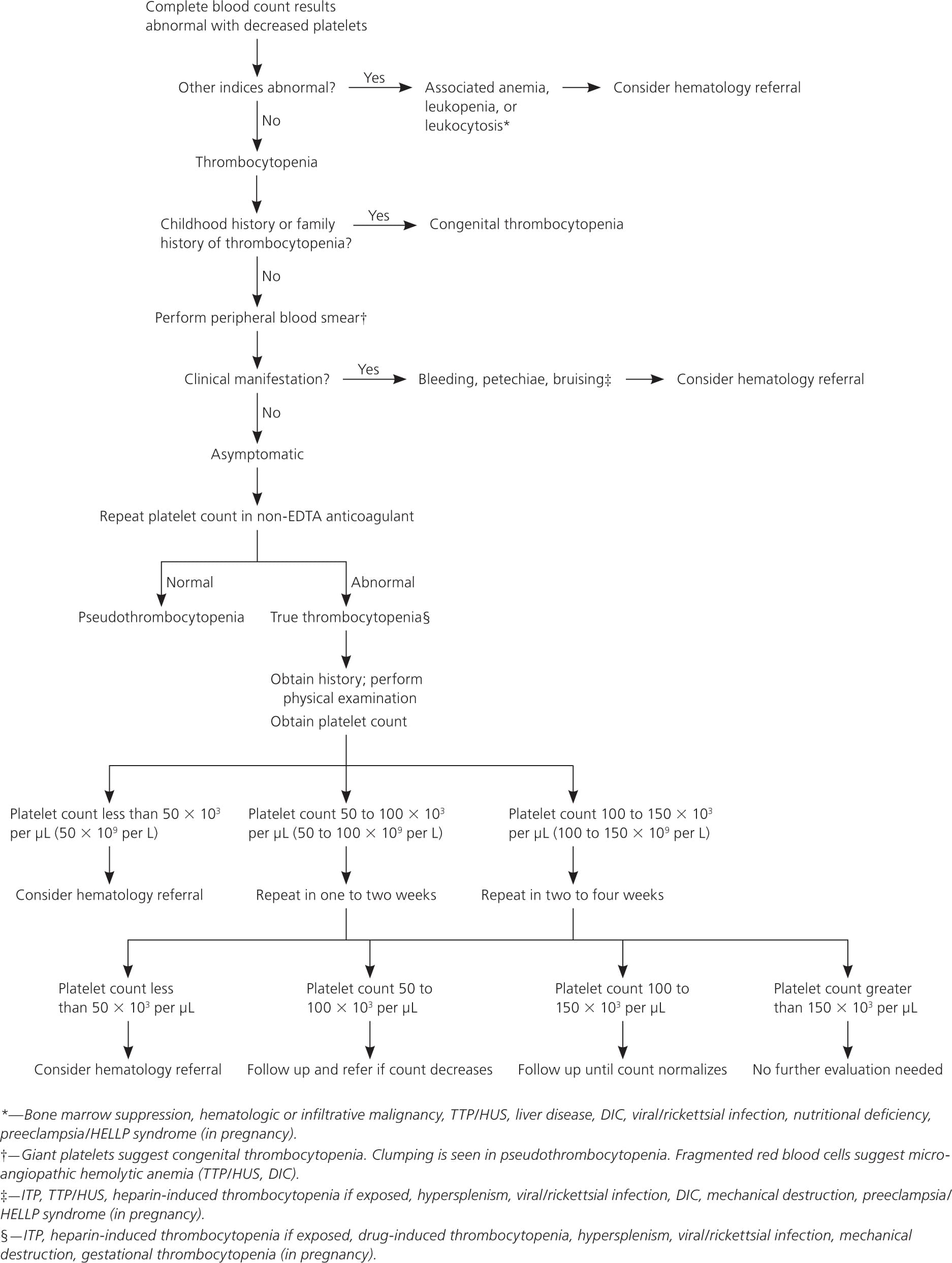
Algorithm for the management of thrombocytopenia. (DIC = disseminated intravascular coagulation; EDTA = ethylenediaminetetraacetic acid; HELLP = hemolysis, elevated liver enzymes, and low platelet count; HUS = hemolytic uremic syndrome; ITP = immune thrombocytopenic purpura; TTP = thrombotic thrombocytopenic purpura.)
Information from reference 4.
Activity Participation and Invasive Procedures
General recommendations for activity participation have been based on historical data from patients with chronic severe thrombocytopenia.39,40 A platelet count greater than 50 × 103 per μL is adequate for hemostasis and is unlikely to be clinically recognized. Patients with a platelet count greater than this level can engage in most activities, but should use caution if participating in contact sports.16,40 Patients with platelet counts less than 10 × 103 per μL should be restricted from contact sports and other potentially traumatic activities. In one study, 42 percent of patients with platelet counts less than 10 × 103 per μL had spontaneous bleeding requiring intervention (e.g., nasal packing) compared with 6 percent of patients with counts between 10 and 30 × 103 per μL.39
Most surgical and invasive procedures can be performed safely in patients with platelet counts greater than 50 × 103 per μL. Other procedures, such as bone marrow biopsy, bronchoscopy, and endoscopy, can be completed safely in patients with platelet counts greater than 20 × 103 per μL, provided that no other bleeding abnormalities are noted.41,42 The American College of Obstetricians and Gynecologists recommendations state that epidural anesthesia in pregnancy is safe in patients with platelet counts between 50 and 100 × 103 per μL.10
Data Sources: The authors searched OvidSP, UpToDate, American Academy of Family Physicians, American College of Obstetricians and Gynecologists, American Society of Clinical Oncology Practice Guidelines, Hematology/Oncology Clinics or North America, Harrison's Principles of Internal Medicine (17th edition), Agency of Healthcare Research and Quality Evidence Reports, National Guidelines Clearinghouse, Cochrane Database of Systematic Reviews, and the Centre for Reviews and Dissemination. Key words included: thrombocytopenia, review, treatment, idiopathic (immune) thrombocytopenic purpura, thrombocytopenia in pregnancy, thrombotic thrombocytopenic purpura, heparin-induced thrombocytopenia, hemolytic uremic syndrome, preeclampsia, HELLP syndrome, gestational thrombocytopenia, pseudothrombocytopenia, myelodysplastic syndrome, microangiopathic hemoglobinopathies, drug-induced thrombocytopenia, platelet transfusion, inherited thrombocytopenias, antiphospholipid antibodies, and viral infections. Information published between 1996 and June 2010 was included. Original search dates: April 5 and June 16, 2010. Final search date: November 23, 2011.
