Hirschsprung’s disease (congenital megacolon) is caused by the failed migration of colonic ganglion cells during gestation. Varying lengths of the distal colon are unable to relax, causing functional colonic obstruction. Hirschsprung’s disease most commonly involves the rectosigmoid region of the colon but can affect the entire colon and, rarely, the small intestine. The disease usually presents in infancy, although some patients present with persistent, severe constipation later in life. Symptoms in infants include difficult bowel movements, poor feeding, poor weight gain, and progressive abdominal distention. Early diagnosis is important to prevent complications (e.g., enterocolitis, colonic rupture). A rectal suction biopsy can detect hypertrophic nerve trunks and the absence of ganglion cells in the colonic submucosa, confirming the diagnosis. Up to one third of patients develop Hirschsprung’s-associated enterocolitis, a significant cause of mortality. Patients should be monitored closely for enterocolitis for years after surgical treatment of Hirschsprung’s disease. With proper treatment, most patients will not have long-term adverse effects and can live normally.
Hirschsprung’s disease occurs in one out of 5,000 births.1 The disease is caused by the failure of ganglion cells to migrate cephalocaudally through the neural crest during weeks four to 12 of gestation,2 causing an absence of ganglion cells in all or part of the colon. Varying lengths of the distal colon are unable to relax, causing functional colonic obstruction over time. The aganglionic segment usually begins at the anus and extends proximally.3 Short-segment disease is most common and is confined to the rectosigmoid region of the colon. Long-segment disease extends past this region and can affect the entire colon. Rarely, the small and large intestines are involved.4 Most patients present in infancy, and early diagnosis is important to avoid complications. With proper treatment, most patients live normal adult lives.
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Hirschsprung’s disease should be confirmed using rectal suction biopsy. | C | 17 |
| Serial rectal irrigation should be performed before surgery to help prevent enterocolitis. | C | 6 |
| Surgery is the recommended treatment for patients with Hirschsprung’s disease. | A | 6 |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, see page 1263 orhttps://www.aafp.org/afpsort.xml.
Epidemiology
The cause of Hirschsprung’s disease is multifactorial, and the disease can be familial or develop spontaneously.2 It is more common in boys than girls.1 Approximately 3 to 5 percent of male siblings and 1 percent of female siblings of children with short-segment disease also have the disease.5 However, the risk is substantially higher (12.4 to 33 percent) in siblings of children with total colonic involvement.6
Eight genomes have been associated with Hirschsprung’s disease1,2; however, most cases are not considered familial. Current research is focusing on the RET proto-oncogene on chromosome 10q11.2.2,7 Hirschsprung’s disease associated with this gene has been linked to multiple endocrine neoplasia, type IIA (i.e., medullary carcinoma of the thyroid and adrenal tumors).7 Research on the value of screening for this mutation to determine the risk of multiple endocrine neoplasia, type IIA, is ongoing.3,8,9
Hirschsprung’s disease also can be associated with neurologic, cardiovascular, urologic, and gastrointestinal abnormalities. Down syndrome (trisomy 21) is the most common chromosomal abnormality associated with the disease, accounting for approximately 10 percent of patients.1 Other conditions that have been linked to Hirschsprung’s disease include congenital deafness, hydrocephalus, diverticulum of the bladder, Meckel’s diverticulum, imperforate anus, ventricular septal defect, renal agenesis, cryptorchidism, Waardenburg’s syndrome (pigment defects associated with deafness), neuroblastomas, and Ondine’s curse (primary alveolar hypoventilation).3,4
Presentation
Symptoms range from neonatal intestinal obstruction to chronic progressive constipation in older children (Table 1).5,6 Approximately 80 percent of patients present in the first few months of life with difficult bowel movements, poor feeding, and progressive abdominal distention.5 Up to 90 percent of infants with Hirschsprung’s disease fail to pass meconium in the first 24 hours of life5; however, other causes of this delay (Table 21,2,10) also should be considered.
TABLE 1 Symptoms of Hirschsprung’s Disease
| Infants |
| Bilious vomiting |
| Enterocolitis-associated diarrhea |
| Failure to pass meconium in the first 24 hours of life |
| Infrequent, explosive bowel movements; difficult bowel movements |
| Jaundice |
| Poor feeding |
| Progressive abdominal distention |
| Tight anal sphincter with an empty rectum |
| Older children |
| Absence of soiling or overflow incontinence |
| Chronic progressive constipation, usually with onset in infancy |
| Failure to thrive |
| Fecal impaction |
| Malnutrition |
| Progressive abdominal distention |
TABLE 2 Diagnoses Associated with a Newborn’s Failure to Pass Meconium
| Diagnosis* | Prevalence (incidence per total births) | Findings |
|---|---|---|
| Meconium plug syndrome | One per 500 to 1,000 | Meconium plugs |
| Cystic fibrosis–associated meconium ileus | One per 2,800 | Abdominal distention at birth, cystic fibrosis |
| Hirschsprung’s disease | One per 5,0001,2 | See Table 1 |
| Anorectal malformation | One per 4,000 to 8,000 | Anal fistula or an absent anus |
| Small left colon syndrome | Rare | Transition zone† at the splenic flexure |
| Hypoganglionosis | Rare | Transition zone† |
| Neuronal intestinal dysplasia | Rare | Megacolon, abnormal ganglion cells |
*—Listed in order of prevalence.
†—From the small- to large-diameter bowel; visible on a contrast enema radiograph.
Infrequent, explosive bowel movements caused by functional colonic obstruction are common in infants with Hirschsprung’s disease. Rectal examination may demonstrate a tight anal sphincter and explosive discharge of stool and gas. Although most patients present in infancy and early childhood, some may not have symptoms until later in life.3,11 Common symptoms in older children include chronic progressive constipation, recurrent fecal impaction, failure to thrive, and malnutrition.12 One third of patients with Hirschsprung’s disease present with enterocolitis-related diarrhea rather than constipation.5
Hirschsprung’s disease can be differentiated from functional constipation if the child is younger than 12 months at onset; fails to pass meconium in the first 24 hours of life; or presents with failure to thrive, absence of overflow incontinence or soiling, or a tight anal sphincter with an empty rectum.13 Symptoms may recur after previously resolving with enemas, laxatives, or feeding changes.5
Diagnosis
Imaging can help diagnose Hirschsprung’s disease. A plain abdominal radiograph may show a dilated small bowel or proximal colon. Contrast enema radiographs of the colon commonly are normal for the first three months of life and indefinitely in patients with total colonic disease. After the dilation process begins, the diseased portion of the colon will appear normal and the more proximal colon will be dilated. A “transition zone” (the point where the normal bowel becomes aganglionic) may be visible on a contrast enema radiograph; however, the aganglionic colon will extend beyond this point in about 10 percent of patients.14–16 Figure 1 includes contrast enema radiographs of an infant with Hirschsprung’s disease. Contrast enemas should be avoided in patients with enterocolitis because of the risk of perforation.6 Anal manometry (balloon distention of the rectum) demonstrates the absence of internal anal sphincter relaxation upon rectal distention.3 Contrast enema and anal manometry are similar in sensitivity and specificity.
Figure 1.
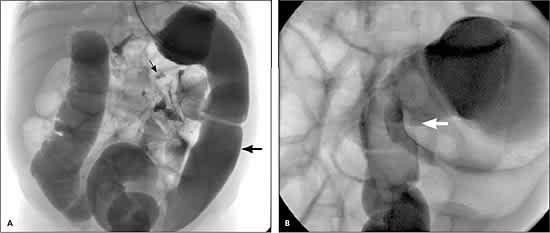
Contrast enema radiographs in an infant with Hirschsprung’s disease. (A) Two weeks of age. Note the dilated small bowel (small arrow) and the normal-appearing colon (large arrow). (B) Four months of age. Note the transition zone in the rectosigmoid region where the normal bowel becomes aganglionic (arrow).
The diagnosis can be confirmed with a rectal suction biopsy, which should show the absence of ganglion cells and the presence of hypertrophic nerve trunks.12,17 Patients typically are referred to a pediatric surgeon or gastroenterologist for biopsy; however, family physicians should be familiar with the procedure to evaluate the outcome of surgery and determine appropriate follow-up. The biopsy site should at least be 0.6 in (1.5 cm) above the dentate line because the distal rectum normally does not have ganglion cells.18 If no hypertrophic nerve trunks are found, a full-thickness biopsy may be indicated.
Treatment
After Hirschsprung’s disease is diagnosed, surgery usually is needed.6 Physicians should have a general knowledge of common procedures to help facilitate communication between the surgeon and the patient’s family. Before surgery, serial rectal irrigation helps decompress the bowel and prevent enterocolitis.6 In otherwise healthy newborns with undistended colons and short-segment Hirschsprung’s disease, the definitive ileoanal pull-through anastomosis can be performed.6,14,19,20 If the child has Hirschsprung’s-associated enterocolitis or a significantly dilated colon, a colostomy can be placed for several months while the child recovers15; the pull-through procedure usually is performed four to six months after colostomy placement.
There are several pull-through techniques, with complication rates ranging from 4 to 16 percent. Swenson’s operation involves removing the rectum, pulling the healthy ganglionated colon through, and connecting it to the anus.5 Newer techniques (e.g., Duhamel operation, Soave operation) help preserve the intricate nerve supply to the rectum and bladder.21 Dilations of the anastomosis are necessary for several months after the Soave operation to prevent stricture formation; the patient’s parents can do this at home. All of these procedures have high success rates, and morbidity is minimal.5,22
Some surgeons perform a one-stage transanal Soave operation in newborns with short-segment disease, eliminating the need for an abdominal incision and colostomy.23 Complication rates have been similar to the more invasive Soave operation; however, short follow-up periods have limited outcome studies of this approach.11,14,21,22
Follow-up
In addition to making an early diagnosis and a prompt referral for treatment, the family physician’s role should include monitoring for postoperative complications and offering counseling and self-help resources to the patient’s family.
COMPLICATIONS
Most patients treated for Hirschsprung’s disease do not have complications. However, up to 10 percent may have constipation, and less than 1 percent may have fecal incontinence.6 Enterocolitis and colonic rupture are the most serious complications associated with the disease and are the most common causes of Hirschsprung’s-related mortality. Enterocolitis occurs in 17 to 50 percent of infants with Hirschsprung’s disease and most commonly is caused by intestinal obstruction and residual aganglionic bowel.5,6 Infants should continue to be monitored closely for enterocolitis many years after corrective surgery because the infection has been reported to occur up to 10 years later. However, most postoperative enterocolitis cases occur within the first two years of ileoanal pull-through anastomosis.6
Early symptoms of enterocolitis (Table 35,6) in patients with Hirschsprung’s disease include abdominal distention; foul-smelling, watery diarrhea; lethargy; and poor feeding. Treatment with rectal irrigation several times per day and antibiotics usually is effective. Oral metronidazole (Flagyl) can be used with rectal irrigation in patients with milder disease.6 More serious disease should be treated intravenously with broad-spectrum antibiotics and rectal irrigation. Rectal irrigation is performed by pushing normal saline into the colon through a rubber catheter; this allows for discharge of gas and stool. The saline (10 to 15 mL per kg) is pushed through the tube, and it is allowed to empty at 10– to 15–mL increments as the catheter is advanced gently.
TABLE 3 Symptoms of Enterocolitis Associated with Hirschsprung’s Disease
COUNSELING
After diagnosis and surgery, physicians should counsel the patient’s family about the importance of a high-fiber diet because constipation and bowel stasis are thought to increase the risk of enterocolitis.23,24 In addition, physicians should consider the conditions that are associated with Hirschsprung’s disease and counsel parents on the risk of the disease in the patient’s siblings. The patient information handout following this article includes a list of resources for more information about Hirschsprung’s disease and support groups.
