Systemic sclerosis (systemic scleroderma) is a chronic connective tissue disease of unknown etiology that causes widespread microvascular damage and excessive deposition of collagen in the skin and internal organs. Raynaud phenomenon and scleroderma (hardening of the skin) are hallmarks of the disease. The typical patient is a young or middle-age woman with a history of Raynaud phenomenon who presents with skin induration and internal organ dysfunction. Clinical evaluation and laboratory testing, along with pulmonary function testing, Doppler echocardiography, and high-resolution computed tomography of the chest, establish the diagnosis and detect visceral involvement. Patients with systemic sclerosis can be classified into two distinct clinical subsets with different patterns of skin and internal organ involvement, autoantibody production, and survival. Prognosis is determined by the degree of internal organ involvement. Although no disease-modifying therapy has been proven effective, complications of systemic sclerosis are treatable, and interventions for organ-specific manifestations have improved substantially. Medications (e.g., calcium channel blockers and angiotensin-II receptor blockers for Raynaud phenomenon, appropriate treatments for gastroesophageal reflux disease) and lifestyle modifications can help prevent complications, such as digital ulcers and Barrett esophagus. Endothelin-1 receptor blockers and phosphodiesterase-5 inhibitors improve pulmonary arterial hypertension. The risk of renal damage from scleroderma renal crisis can be lessened by early detection, prompt initiation of angiotensin-converting enzyme inhibitor therapy, and avoidance of high-dose corticosteroids. Optimal patient care includes an integrated, multidisciplinary approach to promptly and effectively recognize, evaluate, and manage complications and limit end-organ dysfunction.
Systemic sclerosis (systemic scleroderma) is a connective tissue disease associated with autoimmunity, vasculopathy, and fibrosis. The annual incidence is estimated to be 10 to 20 cases per 1 million persons,1 whereas the prevalence is four to 253 cases per 1 million persons.2 Raynaud phenomenon and scleroderma (hardening of the skin) are the clinical hallmarks of the disease. Pulmonary fibrosis and pulmonary arterial hypertension are the leading causes of death.3
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Patients with significant internal organ involvement are often asymptomatic until the late stages of systemic sclerosis; therefore, routine monitoring for underlying disease is essential after the initial diagnosis. | C | 11, 12 |
| Doppler echocardiography, pulmonary function testing, and high-resolution computed tomography of the chest should be performed at diagnosis of systemic sclerosis and at regular intervals thereafter. | C | 7, 8 |
| Treating active interstitial lung disease with oral cyclophosphamide (Cytoxan) for one year modestly improves lung function, dyspnea, skin thickening, and health-related quality of life in patients with systemic sclerosis. | B | 25, 27 |
| Initiation and continuation of angiotensin-converting enzyme inhibitors are recommended in patients with scleroderma renal crisis, even in the presence of elevated creatinine levels. | B | 11, 16 |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.xml.
Epidemiology and Classification
Patients who have systemic sclerosis can be classified into distinct clinical subsets with different patterns of skin and internal organ involvement, autoantibody production, and patient survival.4 The most common subsets are limited cutaneous (approximately 60 percent of patients with systemic sclerosis) and diffuse cutaneous (approximately 35 percent of patients with systemic sclerosis). Table 1 includes the clinical features of limited and diffuse cutaneous systemic sclerosis, and Table 23,4 presents the clinical associations between subtypes and autoantibodies. The limited cutaneous subset is diagnosed when skin thickening is limited to areas distal to the elbows and knees. CREST (calcinosis cutis, Raynaud phenomenon, esophageal dysfunction, sclerodactyly, telangiectasia) syndrome is a variant of limited cutaneous systemic sclerosis. Systemic sclerosis sine scleroderma is a less common subset (approximately 5 percent of patients with systemic sclerosis) that is associated with the characteristic internal organ manifestations of the disease without skin thickening.
Table 1 Distinguishing Clinical Features of Limited Cutaneous and Diffuse Cutaneous Systemic Sclerosis
| Feature | Limited cutaneous | Diffuse cutaneous |
|---|---|---|
| Skin fibrosis | Areas distal to the elbows and knees; may affect the face | Areas proximal or distal to the elbows and knees; may affect the face |
| Typical form of lung involvement | Pulmonary arterial hypertension | Interstitial lung disease |
| Characteristic visceral organ Involvement | Severe gastroesophageal reflux disease and Raynaud phenomenon | Scleroderma renal crisis |
| Physical examination Findings | Telangiectasia, calcinosis cutis, sclerodactyly, digital ischemic complications | Tendon friction rubs, pigment changes |
Table 2 Clinical Associations Between Systemic Sclerosis Subtypes and Scleroderma-Specific Autoantibodies
| Autoantibody | Subtype (percentage with subtype and autoantibody) | Clinical associations |
|---|---|---|
| Antinuclear antibody | Limited cutaneous and diffuse cutaneous (95 percent [nucleolar pattern is most specific]) | Pulmonary arterial hypertension |
| Interstitial lung disease | ||
| Anticentromere antibody | Limited cutaneous (60 to 80 percent) | Pulmonary arterial hypertension |
| Diffuse cutaneous (2 to 5 percent) | Digital ulcerations or digital loss | |
| Antitopoisomerase-1 antibody (anti-Scl-70) | Diffuse cutaneous (20 to 40 percent) | Rapidly progressive skin thickening |
| Scleroderma renal crisis | ||
| Pulmonary fibrosis |
Localized forms of scleroderma, such as linear scleroderma and morphea, primarily affect children and, in contrast to systemic sclerosis, are not associated with Raynaud phenomenon or significant internal organ manifestations. Scleroderma mimics are uncommon conditions that are associated with skin induration, but lack Raynaud phenomenon, internal organ involvement, and autoantibodies. Other diseases, such as mixed or undifferentiated connective tissue disease and overlap syndrome, should be considered before establishing a diagnosis (Table 3).
Table 3 Scleroderma Spectrum Disorders
| Disorder | Variants | |
|---|---|---|
| Diffuse cutaneous systemic sclerosis | — | |
| Limited cutaneous systemic sclerosis | CREST syndrome | |
| Systemic sclerosis sine scleroderma | — | |
| Localized scleroderma | Linear scleroderma | |
| En coup de saber | ||
| Morphea | ||
| Generalized | ||
| Plaque | ||
| Mixed connective tissue disease | Features of systemic sclerosis, polymyositis, and SLE | |
| Overlap syndromes | Systemic sclerosis plus polymyositis, rheumatoid arthritis, or SLE | |
| Scleroderma mimics | Amyloidosis | |
| Chronic graft-versus-host disease | ||
| Diffuse fasciitis with eosinophilia | ||
| Eosinophilia-myalgia syndrome | ||
| Nephrogenic fibrosing dermopathy | ||
| Paraneoplastic syndromes | ||
| Scleredema | ||
| Scleromyxedema (papular mucinosis) | ||
| Toxic oil syndrome | ||
| Undifferentiated connective tissue disease | Multiple nonspecific, serologic or clinical abnormalities that do not meet ACR criteria for rheumatic disease | |
ACR = American College of Rheumatology; CREST = calcinosis cutis, Raynaud phenomenon, esophageal dysfunction, sclerodactyly, telangiectasia; SLE = systemic lupus erythematosus.
Clinical Presentation
A systemic sclerosis diagnosis is based on clinical findings, which have substantial heterogeneity and varying manifestations. The classic clinical presentation is a young or middle-age woman with Raynaud phenomenon and skin changes accompanied by musculoskeletal discomfort and gastrointestinal symptoms. Table 4 summarizes the systemic manifestations of the disease.
Table 4 System-Specific Involvement of Systemic Sclerosis
| System | Manifestation | History and physical examination findings |
|---|---|---|
| Cardiovascular | Abnormal cardiac conduction | — |
| Congestive heart failure | Edema, extra heart sound (S3) | |
| Diastolic dysfunction (secondary to left ventricular fibrosis) | ||
| Pericardial effusion | ||
| Digital ischemic changes | Abnormal capillaries on the nail fold | |
| Acro-osteolysis | ||
| Digital pitting or ulceration | ||
| Pterygium inversus unguis (i.e., distal nail bed adherence to the ventral surface of the nail plate) | ||
| Raynaud phenomenon | Color changes in the fingers precipitated by exposure to cold Temperature or emotional stress: white (vasospasm), blue-purple (ischemia), and red (hyperemia) | |
| Gastrointestinal | Bacterial overgrowth | Anemia |
| Barrett esophagus or strictures | — | |
| Gastric antral vascular ectasia (watermelon stomach) | Anemia | |
| Gastrointestinal bleeding | ||
| Gastroesophageal reflux disease | Chronic cough | |
| Dental erosions | ||
| Dysphagia | ||
| Halitosis | ||
| Pharyngitis | ||
| Intestinal malabsorption | Wasting, diarrhea | |
| Pseudo-obstruction | Obstructive symptoms | |
| Genitourinary | Sexual dysfunction | Dyspareunia, impotence |
| Musculoskeletal | Flexion contractures | Prayer or steeple sign (inability to directly bring hands together because fingers will not fully extend) |
| Muscle atrophy (secondary to myositis [overlap syndrome] or deconditioning) | Weakness | |
| Puffy hands | Diffusely swollen hands without synovitis | |
| Inability to make a tight fist | ||
| Tendon friction rubs | Palpable or audible rubs with active flexion or extension of fingers, wrists, knees, or ankles | |
| Pulmonary | Interstitial lung disease | Basilar, course crackles |
| Cough | ||
| Dyspnea on exertion | ||
| Pulmonary arterial hypertension | Dyspnea on exertion | |
| Extra heart sound (right-sided S3) | ||
| Fixed splitting of S2 | ||
| Right ventricular heave | ||
| Syncope | ||
| Renal | Renal crisis | Abnormal funduscopy examination findings |
| Hypertension | ||
| Schistocytes on peripheral smear | ||
| Skin | Calcinosis | Calcium deposits along extensor tendons and on digits |
| Hyper- or hypopigmentation | Tanned skin on sun-exposed and unexposed areas or loss of pigmentation | |
| Pruritus | Excoriations, scabbing | |
| Telangiectasias | Matte-like vascular abnormalities that blanche on palpation | |
| Thickened skin | Reduced oral aperture | |
| Sclerodactyly | ||
| Tight skin |
S2 = second heart sound; S3 = third heart sound.
RAYNAUD PHENOMENON
Cold-induced Raynaud phenomenon is the most common manifestation of systemic sclerosis, occurring in more than 95 percent of patients. Patients' fingers may change from white (vasospasm) to blue-purple (ischemia) to red (hyperemia); this is precipitated by exposure to cold temperature or emotional stress. Idiopathic or primary Raynaud phenomenon typically occurs in female adolescents, and is not associated with ischemic complications. In contrast, secondary Raynaud phenomenon tends to occur later in life and often leads to tissue damage. Table 5 presents the characteristics of primary and secondary Raynaud phenomenon. Physical findings of secondary Raynaud phenomenon include cyanosis and signs of ischemic damage to the fingers, such as digital pitting (Figure 1A), visible capillaries on the nail bed, ischemic ulcerations (Figure 1B), and pterygium inversus unguis (i.e., distal nail bed adherence to the ventral surface of the nail plate).
Table 5 Characteristics of Primary and Secondary Raynaud Phenomenon
| Characteristic | Primary | Secondary |
|---|---|---|
| Sex of patient | Female | Male or female |
| Age of onset | Adolescence | Adulthood (typically) |
| Symptom severity | Mild to moderate | Moderate to severe |
| Physical examination findings | Normal | Abnormal capillaries on the nail fold (capillaries best visualized using an otoscope); digital pitting or ulcerations; pterygium inversus unguis (i.e., distal nail bed adherence to the ventral surface of the nail plate) |
| Abnormal laboratory findings | None or low-titer ANAs | Low-to-high titer ANAs |
| Incidence of ischemic complications | Rare | Common |
ANA = antinuclear antibodies.
Figure 1.
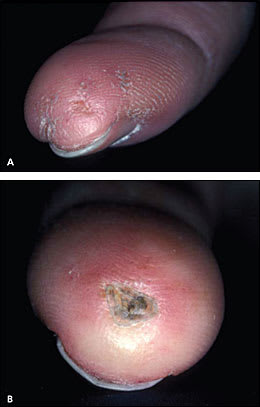
Dermatologic signs of vascular abnormalities in a patient with systemic sclerosis. (A) Digital pitting. (B) Healing digital ulcer.
SKIN MANIFESTATIONS
The degree of skin thickening depends on the subtype and duration of disease. Early in the disease, diffuse swelling of the fingers and hands (Figure 2A) may precede skin thickening and lead to an initial undifferentiated arthritis diagnosis. Other early dermatologic changes include shiny skin (Figure 2B) or pigment changes (Online Figure A1). As the skin thickens on the fingers (sclerodactyly), hands and forearms (limited cutaneous systemic sclerosis), or trunk (diffuse cutaneous systemic sclerosis), the systemic sclerosis diagnosis becomes increasingly apparent.
Figure 2.
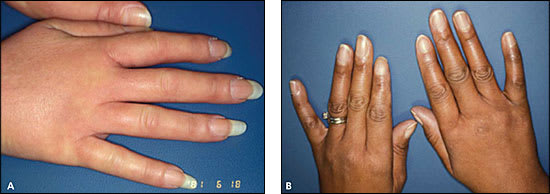
Early dermatologic manifestations of systemic sclerosis. (A) Diffusely puffy hands are a common initial presentation. (B) Shiny skin suggests impending skin thickening.
Facial thickening, which can occur with the limited cutaneous and diffuse cutaneous subsets, often leads to difficulty opening the mouth ( Online Figure A2). Other cutaneous manifestations include hair loss on involved skin; telangiectasia on the face, buccal mucosa, chest, and hands; and calcinosis cutis ( Online Figures B1 and B2). With disease progression, ulcerations over joints and flexion contractures of the fingers, wrists, and elbows may occur.
MUSCULOSKELETAL MANIFESTATIONS
Musculoskeletal involvement is common in early systemic sclerosis and often prompts patients to seek medical evaluation. Puffy hands with arthralgia and myalgia may lead to difficulty making a fist. Palpable or audible friction rubs may be noted over the extensor and flexor tendons of the hands, knees, and ankles. Because friction rubs are highly associated with diffuse cutaneous systemic sclerosis,5 the presence of friction rubs should prompt early diagnosis and screening for characteristic internal organ involvement.
GASTROINTESTINAL MANIFESTATIONS
Symptoms related to gastroesophageal reflux disease (GERD) and dysphagia or changes in bowel habits secondary to intestinal dysmotility are common in patients with early systemic sclerosis. Esophageal disease is virtually universal in patients with the limited cutaneous subset and can cause considerable pathology, even in asymptomatic patients. Bacterial overgrowth in the small bowel (blind loop syndrome) with concomitant nutritional deficiencies (folate and vitamin B12), malabsorption (steatorrhea), and pseudo-obstruction may be a presenting condition, but it is more likely to complicate established disease. Anemia may be a sign of gastric antral vascular ectasia (watermelon stomach). Watermelon stomach refers to the characteristic endoscopic finding of longitudinal rows of sacculated and ectatic mucosal vessels in the antrum of the stomach, which resemble the stripes on a watermelon.
Complications
Internal organ complications are common in patients with systemic sclerosis but are seldom symptomatic until the late stages of the disease; thus, routine screening for internal organ complications is essential.
PULMONARY
Dyspnea is a late manifestation of systemic sclerosis–related lung disease; however, lung involvement is common and is the leading cause of death in patients with systemic sclerosis.3,6 Systemic sclerosis can affect the lung parenchyma (interstitial lung disease) and the pulmonary blood vessels (pulmonary arterial hypertension). Thus, routine screening with pulmonary function tests and Doppler echocardiography in all patients is essential for the early detection of interstitial lung disease and pulmonary arterial hypertension, respectively.7,8
Interstitial Lung Disease
Interstitial lung disease, which is more common with diffuse cutaneous disease, may be preceded by alveolitis that leads to parenchymal fibrosis with destruction of lung architecture and impairment of gas exchange. Interstitial lung disease is suggested when pulmonary function tests reveal restrictive physiology (i.e., a reduction in forced expiratory volume in one second [FEV1] and forced vital capacity [FVC], with a normal FEV1/FVC ratio). Patients with systemic sclerosis who have severe restrictive changes (FVC less than 50 percent) have a 10-year mortality rate of 42 percent.9 Because interstitial lung disease and pulmonary arterial hypertension are both associated with restrictive defects, the FVC/carbon monoxide diffusion in the lung (DLCO) ratio should be calculated. A proportionate reduction in FVC and DLCO, yielding an FVC/DLCO ratio of less than 1.6, suggests interstitial lung disease rather than pulmonary arterial hypertension.9 Reticular or ground-glass opacification in lower lung zones on computed tomography (CT) suggests active alveolitis. Honeycombing, bronchiectasis, and subpleural fibrosis generally occur later in the disease course. Unilateral or upper-lobe abnormalities on high-resolution CT suggest possible infection or malignancy and require further evaluation.
Pulmonary Arterial Hypertension
Elevations in pulmonary arterial pressure are not only secondary to interstitial lung disease and left ventricular dysfunction (secondary pulmonary hypertension), but also to primary obliterative pulmonary arteriopathy (pulmonary arterial hypertension).10 Patients with limited cutaneous systemic sclerosis have the greatest risk of pulmonary arterial hypertension.10 Risk factors for severe pulmonary arterial hypertension include limited cutaneous subset, older age, and elevated pulmonary artery pressures at the initial evaluation.10 Routine screening with Doppler echocardiography and pulmonary function tests may detect pulmonary arterial hypertension before the onset of corpulmonale, when treatment is less effective. However, neither test has sufficient sensitivity or specificity to diagnose or exclude the condition. Other causes of increased pulmonary pressure, such as cardiac valvular disease, embolic disease, obstructive sleep apnea, and hypertensive heart disease, must be excluded. A mean pulmonary artery pressure greater than 25 mm Hg on right heart catheterization is diagnostic for pulmonary arterial hypertension.
RENAL
Before the introduction of angiotensin-converting enzyme (ACE) inhibitors, scleroderma renal crisis was the most fatal complication of systemic sclerosis. Scleroderma renal crisis develops in 3 to 10 percent of all patients with systemic sclerosis and in 10 to 20 percent of those with rapidly progressive diffuse cutaneous systemic sclerosis; the greatest risk occurs within the first three years of the disease.11,12 Other risk factors include high-dose corticosteroid use (greater than 15 mg of prednisone daily),13 the presence of tendon friction rubs, asymptomatic pericardial effusion, new-onset anemia, older age, and pregnancy.11,14 Although antitopoisomerase-1 (anti-Scl-70) antibodies are a marker of diffuse cutaneous systemic sclerosis, their presence does not increase the risk of renal crisis.15 Patients with scleroderma renal crisis characteristically present with sudden-onset accelerated hypertension that is often associated with progressive oliguric renal failure with proteinuria, microangiopathic anemia, and microscopic hematuria. Ten to 15 percent of patients with scleroderma renal crisis are normotensive, but hypertensive when compared with their baseline blood pressure measurements.16 Thus, regular blood pressure monitoring is essential for early detection of scleroderma renal crisis.
CARDIAC
Increasing evidence suggests that systemic sclerosis commonly affects the heart. Cardiac involvement in systemic sclerosis includes myocardial disease, conduction system defects, arrhythmias, or pericardial disease. Scleroderma renal crisis and pulmonary disease can also lead to cardiac dysfunction.17 Single-photon emission CT of asymptomatic patients can detect abnormal microcirculation and vasoreactivity of myocardial vessels.18 Cardiac fibrosis can now be assessed using cardiac magnetic resonance imaging, but no long-term studies have assessed the incidence and outcomes of patients with cardiac fibrosis.
DIFFERENTIAL DIAGNOSIS
The initial evaluation of patients with suspected systemic sclerosis includes a complete blood count; a comprehensive chemistry panel; and serologic studies, including antinuclear, anticentromere, and antitopoisomerase antibodies. Creatine kinase measurements, erythrocyte sedimentation rate, and C-reactive protein measurements may be useful; elevated results suggest myositis, vasculitis, malignancy, or overlap of systemic sclerosis with another autoimmune disease. Table 3 includes scleroderma spectrum disorders.
Treatment
Because of the heterogeneity of systemic sclerosis and potential treatment toxicity, therapy must be individualized to each patient's clinical presentation and needs. No disease-modifying agent has been proven to prevent or reverse fibrosis, although retrospective studies and case series show that d-penicillamine (Cuprimine), mycophenolate mofetil (Cellcept), and cyclophosphamide (Cytoxan) may be effective in some patients. There has been significant improvement in treatments for organ-specific complications (Table 6), especially Raynaud phenomenon, scleroderma renal crisis, and gastrointestinal and pulmonary complications.
Table 6 Treatments for Organ-Specific Complications of Systemic Sclerosis
| Complication | Treatment |
|---|---|
| Raynaud phenomenon | α-Adrenergic blockers |
| Angiotensin-II receptor blockers | |
| Long-acting calcium channel blockers (dihydropyridines) | |
| Pentoxifylline (Trental) | |
| Stellate ganglionic blockades, digital sympathectomy | |
| Skin fibrosis | Immunomodulatory drugs ( |
| Gastroesophageal reflux disease | Antacids |
| Histamine H2 Blockers | |
| Proton pump inhibitors | |
| Intestinal dysmotility or bacterial overgrowth | Antibiotics |
| Correction of nutritional deficiencies | |
| Promotility agents | |
| Pulmonary fibrosis or alveolitis | Immunomodulatory drugs |
| Initial therapy with oral or intravenous cyclophosphamide | |
| Maintenance therapy with azathioprine (Imuran) | |
| Pulmonary arterial hypertension | Diuretics |
| Endothelin-1 receptor inhibitors (bosentan [Tracleer]) | |
| Oxygen | |
| Phosphodiesterase-5 inhibitors (sildenafil [Revatio]) | |
| Prostacyclin analogues (epoprostenol [Flolan], treprostinil [Remodulin], iloprost [Ventavis]) | |
| Warfarin (Coumadin) is sometimes used in patients with recurrent pulmonary thromboembolic disease secondary to pulmonary arterial hypertension | |
| Scleroderma renal crisis | Dialysis |
| Short-acting angiotensin-converting enzyme inhibitors |
note: Complications are listed in descending order of frequency.
RAYNAUD PHENOMENON
Digital amputation because of ischemic complications usually is not necessary if aggressive oral vasodilator therapy is initiated in patients with frequent or severe episodes of Raynaud phenomenon. Commonly used agents include long-acting calcium channel blockers (e.g., nifedipine [Procardia])19 and angiotensin-II receptor blockers (e.g., losartan [Cozaar]).20 There are limited data on the use of phosphodiesterase-5 inhibitors (e.g., sildenafil [Revatio]) for treating secondary Raynaud phenomenon.21 Patients with recurrent ischemic ulcers may benefit from bosentan (Tracleer), an oral endothe-lin-1 receptor inhibitor. In one recent study, patients with ischemic ulcers who were treated with bosentan had a 48 percent reduction in new ulcer formation; however, there was no improvement in existing ulcers.22 Autonomic nervous system modulation with sympatholytic agents (e.g., prazosin [Minipress]) is modestly beneficial and may be associated with adverse effects.23 Patients with symptomatic relief from sympathetic ganglionic blockade may benefit from surgical digital sympathectomy, although there are no controlled trials of its effectiveness.
GASTROINTESTINAL COMPLICATIONS
In addition to therapies to control gastrointestinal symptoms and prevent GERD complications, patients with systemic sclerosis and gastric antral vascular ectasia may require endoscopic laser coagulation to decrease the risk of bleeding. Intestinal pseudo-obstruction often is diagnosed at the time of laparotomy, although conservative nonsurgical management with bowel rest, antibiotics to treat bacterial overgrowth, and judicious use of promotility agents may be effective. Antibiotics, including rifaximin (Xifaxan), and correction of nutritional deficiencies are the mainstays of therapy for intestinal overgrowth.24
PULMONARY COMPLICATIONS
Results from two recent randomized trials suggest that oral or intravenous cyclophosphamide is beneficial in patients with early and progressive interstitial lung disease.25–27 Lung physiology (FVC) and health-related outcomes (dyspnea, skin thickening, quality of life, and function) improved modestly after one year of cyclophosphamide therapy with or without subsequent treatment with azathioprine (Imuran) and prednisone.25–27 It is important to note that although cyclophosphamide has modest benefits for lung function, there is a risk of hemorrhagic cystitis and bladder cancer, bone marrow suppression, infection, infertility, and possibly late hematologic malignancies. Although several small studies suggest a potential role for the immunomodulatory, antifibrotic drug mycophenolate mofetil in the treatment of interstitial lung disease, controlled trials are lacking.28 Oral bosentan, sildenafil, parenteral epoprostenol (Flolan) and treprostinil (Remodulin), and inhaled iloprost (Ventavis) are used to treat symptomatic pulmonary arterial hypertension.29–34 In patients with hypoxemia, continuous oxygen may be needed.
RENAL COMPLICATIONS
All patients with systemic sclerosis should be advised to check their blood pressure at home on a regular basis. Any persistent elevations should prompt a medical evaluation and treatment with ACE inhibitors if scleroderma renal crisis is suspected. ACE inhibitors should be used to control hypertension despite rising serum creatinine levels or the initiation of dialysis because they are essential for preserving and restoring renal function.35
Prognosis
Life expectancy in patients with systemic sclerosis is determined based on the extent and severity of internal organ involvement. Proper management requires regular monitoring and judicious use of organ-specific therapies. Patients may benefit from referral to specialty centers with expertise in treating the various complications of the disease. Self-help resources are available from the Scleroderma Foundation (http://www.scleroderma.org) and the Scleroderma Research Foundation (http://www.sclerodermaresearch.org).