Polycythemia vera is one of three stem-cell–derived myeloid malignancies commonly known as myeloproliferative neoplasms. It is characterized by erythrocytosis, often with associated leukocytosis and thrombocytosis. It has a significant negative impact on overall mortality and morbidity in the form of arterial and venous clots, symptoms of fatigue and pruritus, and conversion to leukemia and myelofibrosis. The World Health Organization's major diagnostic criteria include an elevated hemoglobin or hematocrit level, abnormal results on bone marrow biopsy, and presence of the Janus kinase 2 genetic mutation, which is present in approximately 98% of cases. The only minor criterion is a subnormal erythropoietin level, which helps differentiate polycythemia vera from common causes of secondary erythrocytosis such as smoking, sleep apnea, and testosterone use. First-line treatments, such as low-dose aspirin and goal-directed phlebotomy to a hematocrit level of less than 45% to reduce thrombotic events, improve quality of life and prolong survival. When indicated, cytoreductive therapy, primarily with hydroxyurea, can be added with consideration of second-line agents such as pegylated interferon-alfa, busulfan, and ruxolitinib, depending on the clinical scenario. Smoking cessation and cardiometabolic disease are modifiable risk factors that should be addressed to reduce the risk of thrombosis. Currently, no medications have been shown to cure the disease or to reduce the risk of conversion to leukemia and myelofibrosis.
Polycythemia vera (PV) is one of three common myeloproliferative neoplasms that will likely be encountered during the career of a primary care physician.1 This article summarizes the best, most recent evidence to guide the diagnosis and treatment of PV.
SORT: KEY RECOMMENDATIONS FOR PRACTICE
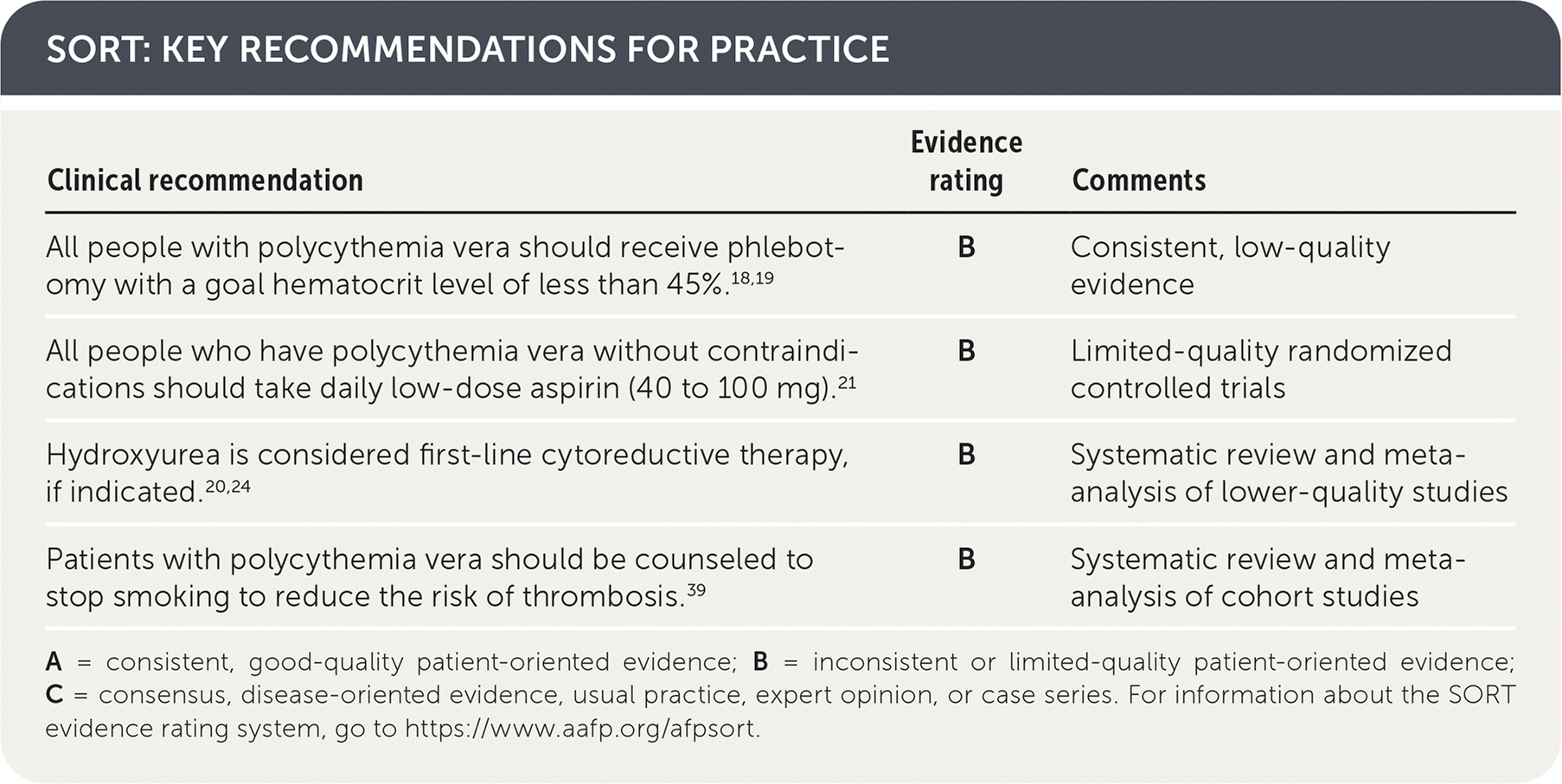
| Clinical recommendation | Evidence rating | Comments |
|---|---|---|
| All people with polycythemia vera should receive phlebotomy with a goal hematocrit level of less than 45%.18,19 | B | Consistent, low-quality evidence |
| All people who have polycythemia vera without contraindications should take daily low-dose aspirin (40 to 100 mg).21 | B | Limited-quality randomized controlled trials |
| Hydroxyurea is considered first-line cytoreductive therapy, if indicated.20,24 | B | Systematic review and meta-analysis of lower-quality studies |
| Patients with polycythemia vera should be counseled to stop smoking to reduce the risk of thrombosis.39 | B | Systematic review and meta-analysis of cohort studies |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.
Epidemiology
- The annual incidence of PV is 0.01 to 2.61 per 100,000 people, and the prevalence is 0.49 to 46.88 per 100,000.2,3
- Median age at diagnosis is 64 years (range = 19 to 95 years), and up to 25% of diagnoses occur before 50 years of age.4,5
- Nonmodifiable risk factors for PV include older age, male sex, White race, and European descent.2,6,7
- Modifiable risk factors for PV include smoking, obesity, hypertension, diabetes mellitus, and hyperlipidemia.8,9
Diagnosis
- Patients who have PV typically present with elevated hemoglobin and hematocrit levels found incidentally or after laboratory evaluation for patients' reported symptoms, physical examination findings, or thrombotic/bleeding events.5,10,11
- The differential diagnosis of PV includes other myeloproliferative neoplasms and secondary erythrocytosis from smoking, sleep apnea, and androgen use and, rarely, erythropoietin-producing tumors (Table 1).12
- Table 2 presents the 2016 World Health Organization's diagnostic criteria for PV.13 A suggested approach to evaluation is summarized in Figure 1.14
TABLE 1. Differential Diagnosis of Polycythemia Vera

| Primary erythrocytosis |
| 2,3-Bisphosphoglycerate deficiency |
| High oxygen-affinity hemoglobin variants |
| Lindau-von Hippel disease |
| Other myeloproliferative neoplasms (essential thrombocytosis, primary myelofibrosis, chronic myelogenous leukemia, myelodysplastic syndrome) |
| Primary familial and congenital polycythemia (EPOR mutation) |
| Secondary erythrocytosis* |
| Cardiopulmonary disease (chronic hypoxia, left-to-right shunt) |
| Erythropoietin-producing tumors (e.g., hepatocellular carcinoma, renal cell carcinoma, hemangioblastoma, pheochromocytoma, uterine leiomyomata) |
| High-altitude habitat |
| Iatrogenic (testosterone, anabolic steroids, erythropoietin, blood doping) |
| Renal disease (renal artery stenosis, hydronephrosis, transplant) |
| Sleep apnea, obesity-hypoventilation syndrome, pickwickian syndrome |
| Smoking, chronic carbon monoxide poisoning |
*—Generally have elevated serum erythropoietin levels.
Information from reference 12.
TABLE 2. 2016 Revised World Health Organization Diagnostic Criteria for Polycythemia Vera

| Must meet all three major criteria or the first two major criteria and the minor criterion |
| Major criteria (1) Hemoglobin > 165 g per L (16.5 g per dL) in men or > 160 g per L (16.0 g per dL) in women or Hematocrit > 49% in men or > 48% in women or Increased red blood cell mass (> 25% above normal) (2) Bone marrow with tri-lineage proliferation with pleomorphic mature megakaryocytes* (3) Presence of Janus kinase 2 mutation |
| Minor criterion Subnormal erythropoietin level |
*—Bone marrow biopsy might not be needed in the presence of a hemoglobin level > 185 g per L (18.5 g per dL) and a hematocrit level of 55.5% in men or a hemoglobin level > 165 g per L (16.5 g per dL) and a hematocrit level of 49.5% in women.
Information from reference 13.
FIGURE 1.
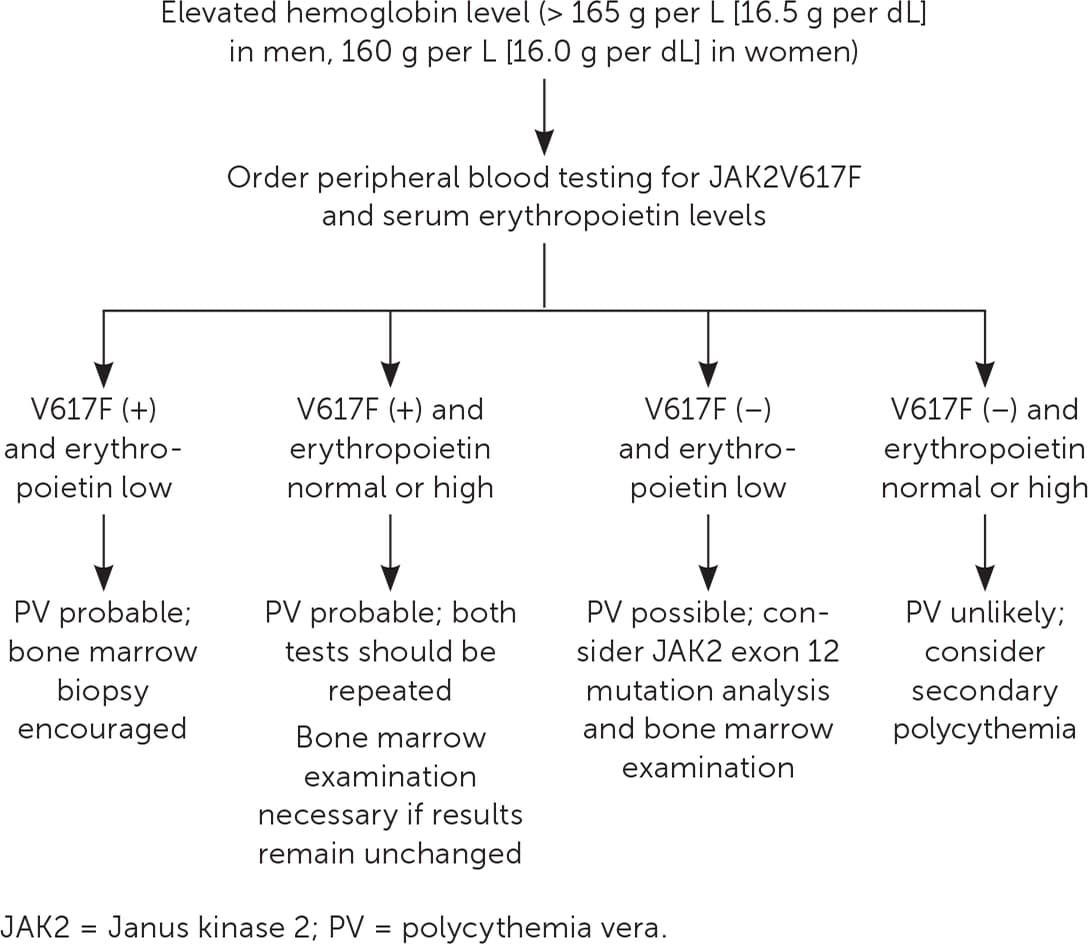
Algorithm for the evaluation of erythrocytosis.
Adapted with permission from Essential Evidence Plus and updated to reflect current diagnostic guidelines. (Essential Evidence Plus. Polycythemia vera. Wiley-Blackwell; 2020. Accessed November 1, 2020. http://www.essentialevidenceplus.com).
SIGNS AND SYMPTOMS
TABLE 3. Laboratory Findings at the Time of Polycythemia Vera Diagnosis
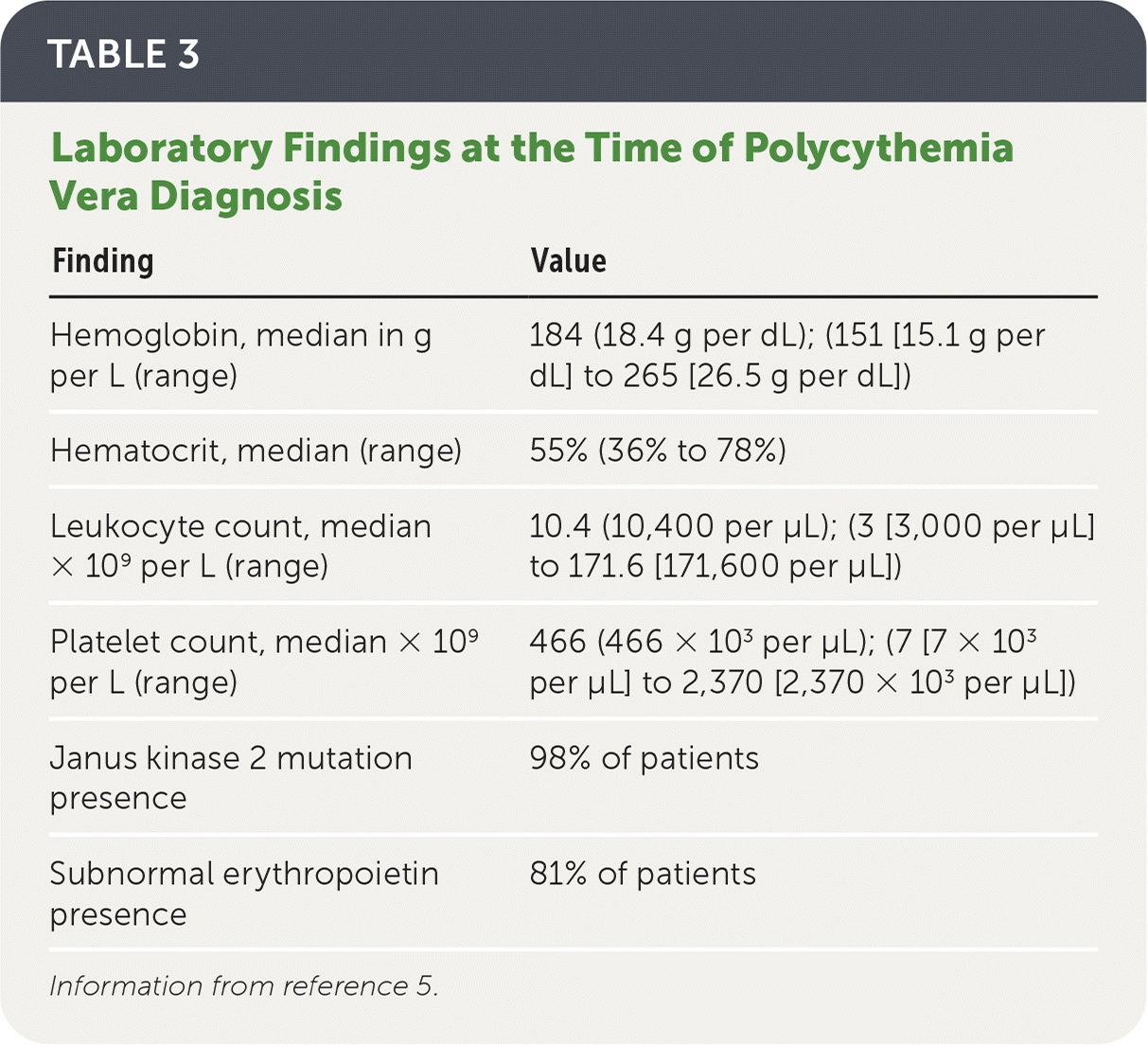
| Finding | Value |
|---|---|
| Hemoglobin, median in g per L (range) | 184 (18.4 g per dL); (151 [15.1 g per dL] to 265 [26.5 g per dL]) |
| Hematocrit, median (range) | 55% (36% to 78%) |
| Leukocyte count, median × 109 per L (range) | 10.4 (10,400 per μL); (3 [3,000 per μL] to 171.6 [171,600 per μL]) |
| Platelet count, median × 109 per L (range) | 466 (466 × 103 per μL); (7 [7 × 103 per μL] to 2,370 [2,370 × 103 per μL]) |
| Janus kinase 2 mutation presence | 98% of patients |
| Subnormal erythropoietin presence | 81% of patients |
Information from reference 5.
TABLE 4. Presenting Features of Polycythemia Vera
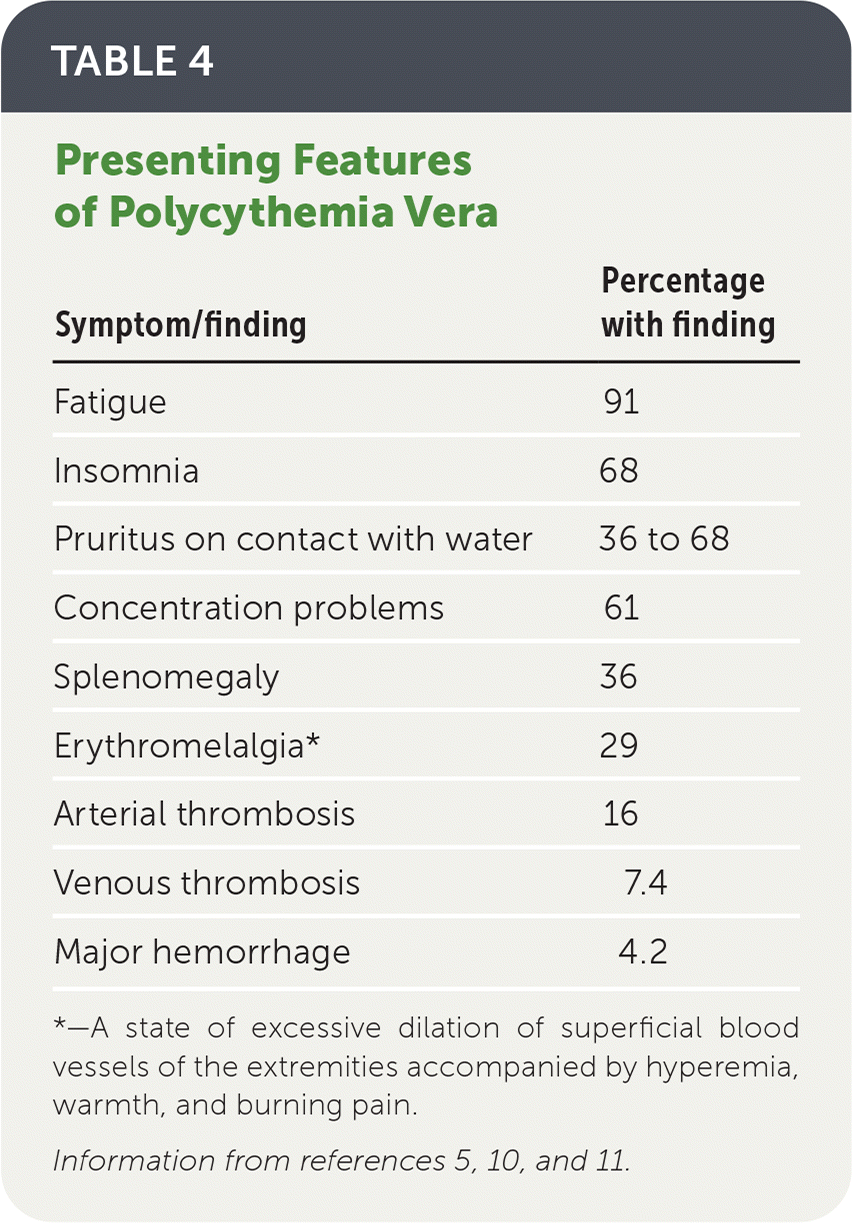
| Symptom/finding | Percentage with finding |
|---|---|
| Fatigue | 91 |
| Insomnia | 68 |
| Pruritus on contact with water | 36 to 68 |
| Concentration problems | 61 |
| Splenomegaly | 36 |
| Erythromelalgia* | 29 |
| Arterial thrombosis | 16 |
| Venous thrombosis | 7.4 |
| Major hemorrhage | 4.2 |
*—A state of excessive dilation of superficial blood vessels of the extremities accompanied by hyperemia, warmth, and burning pain.
DIAGNOSTIC TESTING
- If PV is suspected after an elevated hemoglobin/hematocrit level is found, Janus kinase 2 (JAK2) V617F mutation and erythropoietin testing should be performed.12
- Bone marrow biopsy with fluorescence in situ hybridization testing and karyotyping can be helpful to confirm the diagnosis and to stratify risk.12
Treatment
- The goals of therapy are to improve survival and to improve quality of life by decreasing hematocrit levels, symptom burden, bleeding, and clotting complications.15
- No treatments have been shown to reduce the risk of transformation to leukemia or myelofibrosis. Figure 2 presents a suggested approach to the treatment of PV.12,16,17
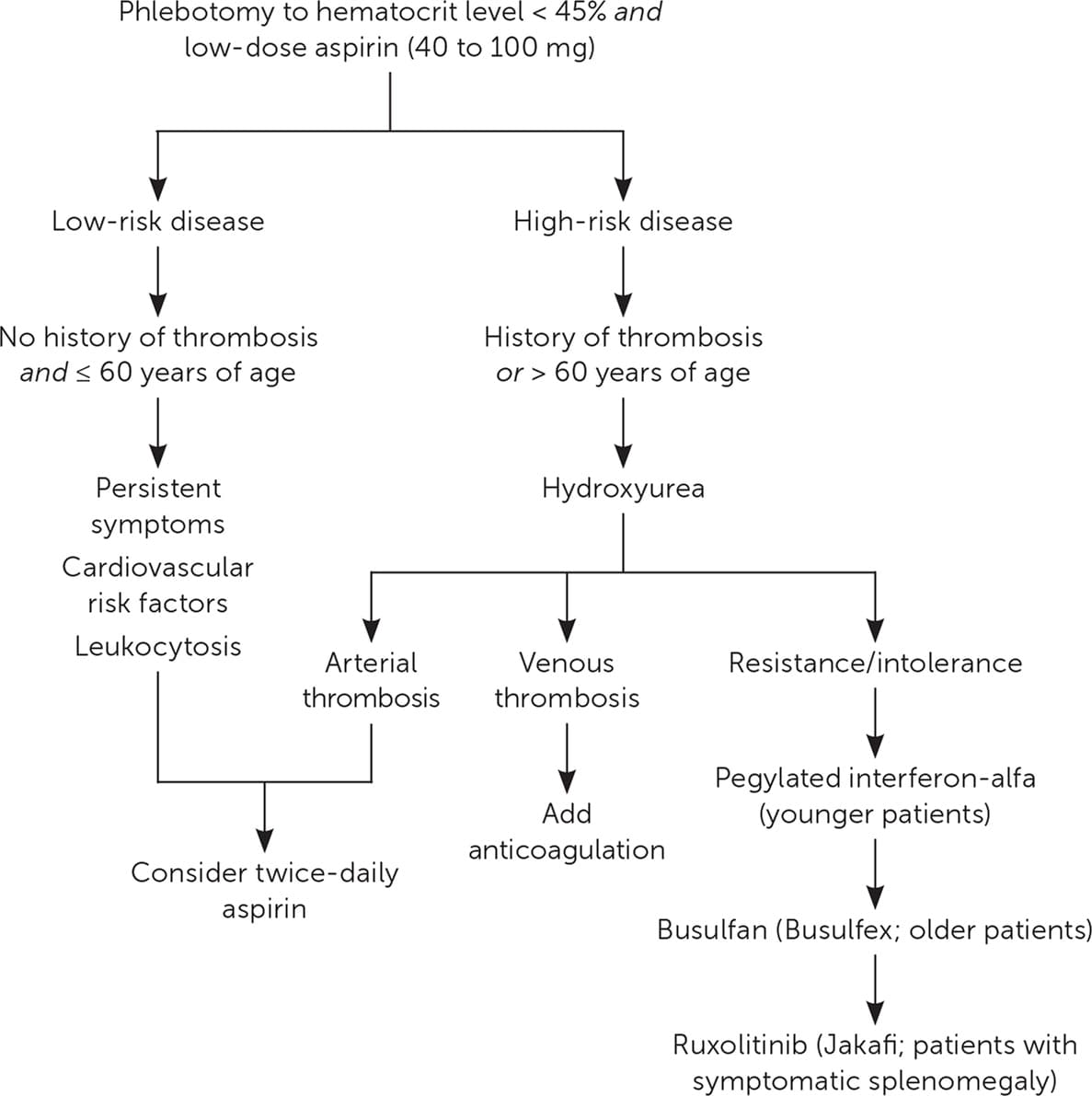
PHLEBOTOMY
- All patients should receive phlebotomy with a goal hematocrit level of less than 45%.18–20
- A randomized controlled trial of 365 patients who had PV with an average age of 65 years showed a decreased incidence of death from cardiovascular events and thrombotic events if phlebotomy was used to keep the hematocrit level at less than 45% vs. 45% to 50% over a median follow-up period of 31 months (1.1 events vs. 4.4 events per 100 person-years; number needed to treat = 30).18
- In a retrospective cohort study, 820 predominantly White patients with Medicare, a median age of 77 years, and a low rate of previous thrombosis (13%) were followed for 2.8 years. The study found that phlebotomy used a median of 2.3 times per year was associated with significantly improved survival compared with no phlebotomy (6.3 years vs. 4.5 years). The risk of thrombosis was also significantly lower with phlebotomy use (29% vs. 46%).19
THROMBOPROPHYLAXIS
- All patients should receive daily low-dose aspirin (40 to 100 mg) in the absence of contraindications.20,21
- A multicenter randomized controlled trial of 518 patients with a median age of 61 years showed that 100 mg of aspirin used daily over a five-year period significantly reduced the risk of nonfatal myocardial infarction, nonfatal stroke, pulmonary embolism, major venous thrombosis, or death from cardiovascular causes compared with placebo (3.2% vs. 7.9%; number needed to treat = 21 over five years).21
- Low-dose aspirin reduces vasomotor symptoms such as headache, erythromelalgia, and pruritus in conjunction with phlebotomy.22
- Aspirin use should be avoided in people with extreme thrombocytosis (greater than 1,000 × 109 per L [1,000 × 103 per μL]) because of increased risk of bleeding from acquired von Willebrand disease.10
CYTOREDUCTIVE THERAPY
Hydroxyurea
- Hydroxyurea is the first-line agent for cytoreductive therapy in PV.20
- The previously mentioned retrospective cohort study of 820 patients with a median age of 77 years found that use of hydroxyurea was associated with significantly improved survival compared with nonusers (6.0 years vs. 5.3 years). The risk of thrombosis was significantly lower in those using hydroxyurea (27.6% vs. 45.4%).19
- A retrospective analysis of 1,042 patients followed over approximately 30 years compared use of hydroxyurea to phlebotomy only and found that fewer cardiovascular events occurred with hydroxyurea (3.0 vs. 5.8 per 100 person-years).23
- A 2019 systematic review and meta-analysis of 3,236 high-risk patients who had PV and were treated with hydroxyurea found that the incidence of leukemic transformation was 0.4% of persons per year, and the rate of myelofibrosis was 5.0% at five years and 33.7% at 10 years.24
- Up to 24% of patients may be resistant or intolerant to hydroxyurea (Table 525 ), and common adverse effects include anemia, neutropenia, oral ulcers, skin ulcers, hyperpigmentation, and nail changes.1,25
- In patients younger than 40 years, caution with hydroxyurea use is recommended because of leukemogenic concerns with long-term use.26
TABLE 5. European LeukemiaNet Definition of Resistance/Intolerance to Hydroxyurea in Patients with Polycythemia Vera

| Hydroxyurea resistance | Hydroxyurea intolerance |
|---|---|
| (1) Need for phlebotomy to keep hematocrit level < 45% after 3 months of hydroxyurea (at least 2 g per day) or (2) Uncontrolled myeloproliferation (i.e., platelet count of 400 × 109 per L [400 × 103 per μL] and white blood cell count of 10 × 109 per L [10,000 per μL]) after 3 months of hydroxyurea (at least 2 g per day) or (3) Failure to reduce massive* splenomegaly by 50% as measured by palpation or failure to completely relieve symptoms related to splenomegaly after 3 months of hydroxyurea (at least 2 g per day) | (1) Absolute neutrophil count of 1.0 × 109 per L (1,000 per μL) or platelet count of 100 × 109 per L (100 × 103 per μL) or hemoglobin level of 100 g per L (10 g per dL) at the lowest dose of hydroxyurea required to achieve a complete or partial clinicohematologic response† or (2) Presence of leg ulcers or other hydroxyurea-related nonhematologic toxicities, such as mucocutaneous manifestations, gastrointestinal symptoms, pneumonitis, or fever with any dose of hydroxyurea |
*—Organ extending by 10 cm from the costal margin.
†—Complete response is defined as hematocrit level of less than 45% without phlebotomy, platelet count of 400 × 109 per L, white blood cell count of 10 × 109 per L, and no disease-related symptoms. Partial response is defined as hematocrit level of less than 45% without phlebotomy or response in 3 or more of the other criteria.
Information from reference 25.
Pegylated Interferon-Alfa
- A disease-oriented, uncontrolled study of 40 patients with high-risk PV reported that when pegylated interferon-alfa was used as first-line therapy, complete hematologic response (76% to 95%) and molecular response (18%) could be achieved over a 36-month period.27 The hematologic and molecular responses were defined by a hematocrit level of less than 45% in men and less than 42% in women without phlebotomy, absence of splenomegaly, normal leukocyte and platelet counts, and undetectable JAK2V617F levels.27
- Up to 20% of patients discontinued the drug because of toxicity (e.g., skin toxicities possibly resulting in rash over more than 50% of body surface area or needing urgent treatment), asthenia, and hematologic abnormalities.27
Busulfan (Busulfex)
- A disease-oriented, retrospective study of 19 patients with PV and a median age of 76 years who had hydroxyurea resistance or intolerance (Table 525 ) and were started on busulfan as second-line therapy reported that 75% of patients had complete or partial hematologic response over a median of 3.8 years.28 Adverse effects such as cytopenias caused 15% of patients to discontinue the medication.28
- Busulfan should be avoided in younger patients because of a higher association with leukemogenicity based on observational data.29
Ruxolitinib (Jakafi)
- A disease-oriented, randomized trial was conducted involving patients who were hydroxyurea resistant or intolerant. In these patients, ruxolitinib achieved better hematocrit control and spleen volume improvement (decrease in volume greater than 35%) compared with other single-drug alternatives selected by the investigator, such as hydroxyurea (tolerable doses), pegylated interferon-alfa, pipobroman (not available in the United States), anagrelide (Agrylin), immunomodulators, or no medication (busulfan was prohibited) over a 32-week period (21% with ruxolitinib vs. 0.9% with other single-drug alternatives; P < .001; number needed to treat = 5).30
- Immunosuppressive adverse effects made herpes zoster infection more likely (6% with ruxolitinib vs. 0% with other single-drug alternatives).30
PRURITUS
TREATMENT IN PREGNANCY
- PV during pregnancy is rare, with a prevalence of less than 0.03 per 100,000.36
- A multidisciplinary team, including an obstetrician, maternal-fetal medicine specialist, and hematologist, is recommended.37
- Potentially teratogenic medications should be stopped in men and women at least three months before conception.36,37
- All pregnant patients who have PV should receive low-dose aspirin, avoid iron supplementation in the absence of actual depletion, maintain gestational age-appropriate hematocrit levels, and be treated with enoxaparin (Lovenox) for six weeks postpartum if no contraindications occur.36
- Interferon-alfa is the drug of choice for cytoreductive therapy if needed.36
Follow-up and Monitoring
- Follow-up appointments every three to six months are recommended for routine history, physical examination, and complete blood count.15
Prognosis
- Poor prognostic features include age older than 60 years, history of thrombosis, leukocytosis, high JAK2 burden, abnormal karyotype, and established cardiovascular risk factors such as smoking, hypertension, diabetes, obesity, and hyperlipidemia.5,9,38
- Smoking cessation decreases the risk of thrombosis.39
- Without treatment, death typically occurs within two years, mostly from thrombotic events.40
- Median survival of patients with PV using standard-of-care treatment, including aspirin and hydroxyurea, is 13.5 years.4
- For those diagnosed before 60 years of age, median survival is 24 years.4
- The risk of blast transformation to acute myeloid leukemia or myelodysplastic syndrome over 15 years is 5.5% to 18.7%.4,41
- Progression to myelofibrosis over 15 years is 6% to 14%.4,41 A practical tool to calculate survival has been developed and is shown in Table 6.5
TABLE 6. Risk Factor–Based Survival Prediction in Polycythemia Vera
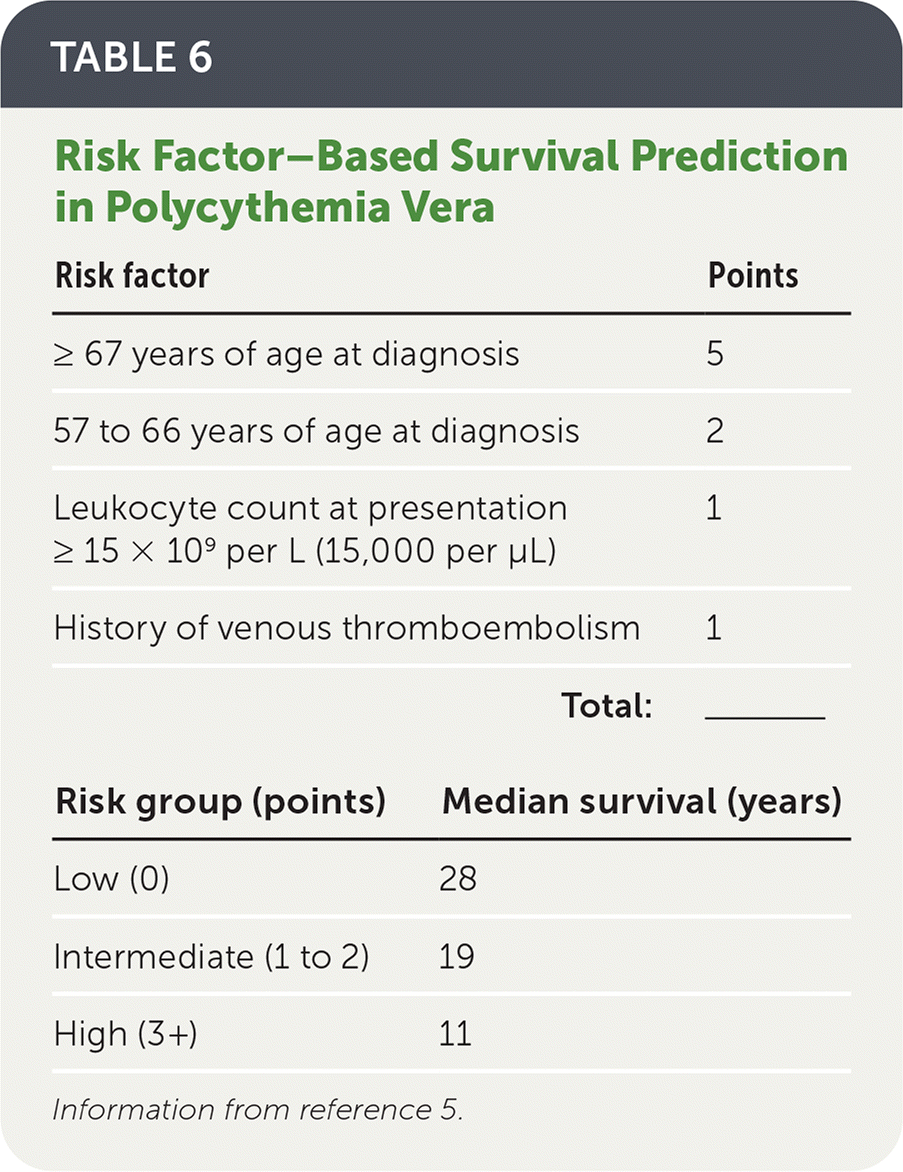
| Risk factor | Points |
|---|---|
| ≥ 67 years of age at diagnosis | 5 |
| 57 to 66 years of age at diagnosis | 2 |
| Leukocyte count at presentation ≥ 15 × 109 per L (15,000 per μL) | 1 |
| History of venous thromboembolism | 1 |
| Total: | ______ |
| Risk group (points) | Median survival (years) |
| Low (0) | 28 |
| Intermediate (1 to 2) | 19 |
| High (3+) | 11 |
Information from reference 5.
This article updates a previous article on this topic by Stuart and Viera.1
Data Sources: We searched Essential Evidence Plus, Cochrane Database of Systematic Reviews, and PubMed with the key words polycythemia vera, myeloproliferative neoplasms, epidemiology, diagnosis, treatment, therapy, prognosis, risk stratification, management, pregnancy, randomized controlled trial, meta-analysis, systematic review, aspirin, hydroxyurea, phlebotomy, interferon-alpha, busulfan, ruxolitinib, and JAK2 kinase inhibitors. Search dates: October 2019 and January 2021.