Sarcoidosis is a multisystemic disorder of unknown etiology that most commonly affects adults between 20 and 40 years of age. Patients with sarcoidosis frequently present with bilateral hilar lymphadenopathy and pulmonary infiltration, and often with ocular and skin lesions. The diagnosis is established when clinical and radiographic findings are supported by histologic evidence of non-caseating epithelioid cell granulomas found on tissue biopsy. Diagnosis of sarcoidosis requires exclusion of other causes of granuloma formation. Sarcoidosis is also characterized by distinctive laboratory abnormalities, including hyperglobulinemia, an elevated serum angiotensin converting enzyme level, evidence of depressed cellular immunity manifested by cutaneous anergy and, occasionally, hypercalcemia and hypercalciuria. Glucocorticoids remain the mainstay of therapy when treatment is required, although other anti-inflammatory agents are being used increasingly often.
Sarcoidosis is a multisystemic disease of unknown etiology that may affect any organ or system in the body. It most commonly affects adults of both sexes between the ages of 20 and 40, but it may occur at any age. Two thirds of patients with sarcoidosis are less than 40 years old at the time of diagnosis. The prevalence of sarcoidosis is slightly higher in females than in males, but these rates vary widely in different countries and populations. Sarcoidosis was formerly thought to be 10 times more common in blacks than in whites, but more recent data indicates a 3.2:1 ratio.1 Disease severity is somewhat worse in blacks.2
In the United States, the incidence of sarcoidosis has been reported to be from one in 2,500 to one in 10,000. It is a very common disorder in the black population of the Caribbean region, particularly when migration to Europe occurs. However, throughout the world, 79 percent of patients with sarcoidosis are white. Sarcoidosis occurs frequently in the white population of Scandinavia; in Sweden, the incidence of sarcoidosis is 64 per 100,000 persons.2 It is also frequently diagnosed in Ireland and throughout Northern Europe. In Japan, familial and environmental factors have been reported, and the human leukocyte antigen subgroup HLA-DR52w has frequently been noted there3 but is not a risk factor for sarcoidosis.
It has been hypothesized that sarcoidosis is the result of various antigens causing T-cell activation in a genetically predisposed host. The increased number of “helper” T cells attracts monocytes, and granuloma formation occurs. B-cell activation also occurs, increasing the production of immunoglobulin.4 Serum angiotensin converting enzymes and active vitamin D may play roles as mediators that increase the inflammatory response.1,3
Sarcoidosis with pulmonary involvement is primarily a restrictive lung disease with abnormal gas exchange but may occasionally present with an obstructive pattern. The pathophysiology of sarcoidosis is similar to that of other interstitial lung diseases, with main features including decreased lung compliance, and decreased lung volumes and capacities. Diminished carbon monoxide diffusing capacity and an increased alveolar-arterial oxygen gradient also occur.5 The latter may cause hypoxemia at rest that worsens with exercise.
Illustrative Case
A 28-year-old white man, previously in good health, presented with a two-week history of cough that produced small amounts of yellow sputum and a one-day history of “streak” hemoptysis. He had a 10–pack-year history of smoking. He denied chest pain, exposure to tuberculosis, hoarseness, recent travel or familial lung disease. His wife had recently been treated for mycoplasmal pneumonia. He also complained of mild dyspnea of one month's duration and recent malaise but had no significant weight changes. His past medical history and review of systems were entirely noncontributory.
Physical examination revealed a moderately obese man with normal vital signs and no respiratory distress. Examination of the head, ears, eyes, nose and throat was normal; there was no cervical adenopathy. His cardiovascular examination was normal. Examination of the chest revealed harsh breath sounds throughout all fields, but no wheezes were noted. Abdominal, neurologic, skin and extremity examinations were negative for pathology. No peripheral adenopathy was noted.
Initial evaluation included a chest radiograph revealing extensive bilateral hilar adenopathy with a large right paratracheal node, but no infiltrates were noted (Figure 1). A complete blood count showed a normal hemoglobin level (15.2 g per dL [152 g per L]) and hematocrit level (44.6 percent [0.45]), normal red blood cell indices and a white blood cell count of 7,200 mm3 (7.2 × 109 per L) with a normal differential. Cold agglutinins were negative. All serum chemistries, including calcium, renal and liver panels, were normal. Pulmonary function testing showed a forced expiratory volume in one second (FEV1) of 80 percent predicted but was otherwise entirely normal. Arterial blood gases were normal. Results of an electrocardiogram were normal. Skin tests for anergy and tuberculosis were negative (i.e., normal). The patient was referred to a pulmonologist, and a subsequent bronchoscopy revealed a slightly injected right mainstem bronchus; at the same time, mediastinoscopy was performed and biopsies were taken from seven lymph nodes, revealing “non-caseating granulomas” consistent with sarcoidosis. A serum angiotensin converting enzyme test was performed, and levels were found to be 240 nmol per mL per min (4,000 nkat per L). The normal range is 10 to 50 nmol per mL per min (167 to 834 nkat per L).
FIGURE 1.
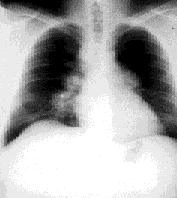
Initial chest radiograph of a patient with sarcoidosis, revealing bilateral hilar adenopathy and a large right paratracheal node.
The patient was advised to stop smoking, but no other therapy was administered. Annual radiographs were performed and, four years later, a radiograph revealed 90 percent clearing of the adenopathy. Results from pulmonary function testing remained normal. The patient's only complaint was “occasional” palpitations, but a thorough cardiac evaluation was unremarkable. No therapy was necessary during the next 10 years, and the patient has remained “disease-free” (Figure 2).
FIGURE 2.
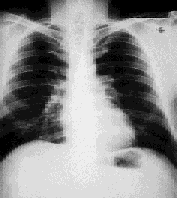
Significant clearing of the hilar adenopathy in the same patient 10 years after the initial chest radiograph.
Clinical Manifestations
Symptoms of sarcoidosis may be caused by a number of factors, including the “mass effect” of the granuloma(s); immune complex vasculitis (as occurs in erythema nodosum); metabolically active granulomas; and fibrotic distortion lasting even after resolution of the granulomatous lesions.
The lungs are the primary target of this disease. About 88 percent of patients with sarcoidosis have lung involvement. It is customary to stage intrathoracic sarcoidosis by comparing current chest radiographs with the chest radiograph taken on initial presentation. Intrathoracic sarcoidosis is divided into four stages. Approximately 8 percent of patients with sarcoidosis present at stage zero. During this stage, the chest radiograph is normal in the presence of multisystem involvement. Results of pulmonary function testing are usually normal, and most patients remit spontaneously. About 51 percent of patients (including the patient described in the illustrative case) present at stage 1. During this stage, chest radiographs show bilateral hilar lymphadenopathy with or without enlarged right paratracheal nodes (Figure 3). Results of pulmonary function tests are usually normal except for a decreased diffusing capacity, but mechanics are normal. Most patients are asymptomatic or have nonpulmonary symptoms. Most patients (70 to 75 percent) remit within two years, and only 10 to 15 percent progress to stage 2.
FIGURE 3.
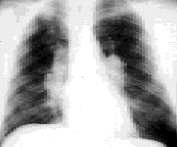
Posteroanterior view of a patient with stage 1 sarcoidosis.
Twenty-nine percent of patients with sarcoidosis present at stage 2. During this stage, chest radiographs show hilar lymphadenopathy associated with diffuse pulmonary infiltration (Figure 4). The signs and symptoms are usually mild in relation to the severity of the abnormalities shown on radiograph. Multiple pulmonary nodules or infiltrates may also be present. Results of pulmonary function testing demonstrate restrictive disease with a decreased diffusing capacity, although obstructive changes resulting from bronchial involvement may also be present. One half of these patients undergo spontaneous remission, but 25 to 30 percent remain at stage 2 or progress to stage 3. In patients with stage 3 sarcoidosis, the chest radiograph shows diffuse pulmonary infiltration without hilar lymphadenopathy (Figure 5). Only about 12 percent of patients present at stage 3. The chest radiograph frequently shows fibrosis with small lung volumes, elevation of the diaphragms and “honeycombing” (fine fibrosis occurring throughout the interstitial lung tissue).
FIGURE 4.
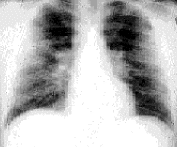
Posteroanterior view of a patient with stage 2 sarcoidosis.
FIGURE 5.
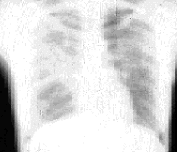
Posteroanterior view of a patient with stage 3 sarcoidosis.
The patient presenting with stage 3 sarcoidosis may have minimal symptoms, (i.e., cough, dyspnea, mild weight loss) or significant problems, including pulmonary hypertension, cor pulmonale and respiratory failure. Many patients in stage 3 have intrinsic restrictive changes on pulmonary function testing but, as a result of bronchial involvement, many also have obstructive changes. Patients at stage 3 usually undergo a chronic course; complications such as pulmonary fibrosis are common and irreversible. Also, at this stage, extrapulmonary findings are more common, especially skin involvement.6 In up to 30 percent of patients at stage 3, sarcoidosis spontaneously remits within two years. Table 1 shows the stages of sarcoidosis and the radiographic findings at the time of diagnosis. Other intrathoracic radiographic findings seen in patients with sarcoidosis include alveolar infiltrates that may appear extensive or patchy, atelectasis, nodular cavitation, pleural thickening, pleural effusions and calcifications.
TABLE 1 Stages of Sarcoidosis
| Stage | Patients presenting at this stage (%) | Findings on chest radiograph | Results of pulmonary function testing | Signs and symptoms | Patients expected to go into remission (%) |
|---|---|---|---|---|---|
| 0 | 8 to 10 | Normal (but with multisystem involvement) | Normal | Varies with system affected | Most remit spontaneously |
| 1 | 51 | Bilateral hilar lymphadenopathy with or without enlarged right paratracheal nodes | Normal, except for decreased diffusing capacity; normal mechanics | Most asymptomatic or with nonpulmonary complaints | 70 to 75% remit within two years; 10 to 15% progress to stage 2 |
| 2 | 29 | Hilar lymphadenopathy with Diffuse pulmonary infiltration; pulmonary nodules may be seen | Usually restrictive changes with decreased diffusing capacity; obstructive changes may be present | Usually mild in relation to the severity of the radiographic findings | 50% spontaneously remit; 25 to 30% persist at stage 2 or progress to stage 3 |
| 3 | 12 | Diffuse pulmonary infiltration, but without hilar lymphadenopathy; fibrosis; small lung volumes; elevated diaphragms; effusions; calcifications; “honeycombing” | Primarily restrictive changes, but with obstructive changes due to bronchial involvement; changes may be severe | Varies: may be minimal (cough, dyspnea, weight loss) to severe (corpulmonale, pulmonary hypertension; may progress to respiratory failure) | 30% spontaneously remit within two years |
Information from reference 2.
A large number of patients (40 to 50 percent) with chronic sarcoidosis have extrapulmonary involvement that can affect almost any tissue. In some patients, it is this extrathoracic sarcoidosis that initially causes the patient to seek medical attention. Table 2 shows the frequency of intrathoracic and extrathoracic manifestations of sarcoidosis.
TABLE 2 Frequency of Extrathoracic and Intrathoracic Manifestations of Sarcoidosis
| Feature | Approximate percentage | ||
|---|---|---|---|
| Intrathoracic | 88 | ||
| Extrapulmonary | 40 to 50 | ||
| Ocular | 27 | ||
| Skin lesions | |||
| Erythema nodosum | 15 to 34 | ||
| Lupus pernio | 4 to 9 | ||
| Other lesions | 14 | ||
| Parotid enlargement | 6 | ||
| Splenomegaly | 10 to 13 | ||
| Hepatomegaly | 10 | ||
| Lymphadenopathy (peripheral) | 27 | ||
| Bone | 3 to 4 | ||
| Heart | 3 to 5 | ||
| Kidney | 1 | ||
| Nervous system | 4 to 5 | ||
| Upper respiratory tract | 2 to 18 | ||
| Hyperglobulinemia | 31 | ||
| Hypercalcemia | 18 | ||
| Anergy | 50 | ||
Approximately one third to one half of patients with chronic sarcoidosis have skin lesions. Skin manifestations of sarcoidosis can be either specific or nonspecific, but most are granulomatous in nature and occur more commonly in black patients. Erythema nodosum is by far the most common skin lesion occurring in patients with sarcoidosis, affecting 15 to 34 percent of patients in most studies (Figure 6). In Britain and Scandinavia, erythema nodosum occurs in 15 to 20 percent of patients. It occurs most often in women of childbearing age and is often associated with pregnancy or lactation, suggesting an etiologic hormonal co-factor. More than 90 percent of patients with sarcoidosis who develop erythema nodosum present with a stage 1 chest radiograph.6 Lofgren's syndrome, which consists of erythema nodosum, fevers, bilateral hilar adenopathy and myalgias, may also occur and is considered an “acute” form of sarcoidosis. Lupus pernio, which consists of persistent violaceous lesions on the nose, cheeks and ears, occurs in 4 to 9 percent of patients with sarcoidosis (Figure 7). It is a manifestation of chronic fibrotic sarcoidosis and is associated with involvement of the upper respiratory tract, advanced pulmonary fibrosis, bone cysts and ocular disease. Other skin lesions include persistent plaques, subcutaneous nodules, ulcerations, maculopapular eruptions, scars and keloids.
FIGURE 6.
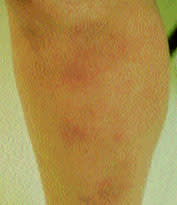
Erythema nodosum on the leg of a patient with sarcoidosis.
FIGURE 7.

Lupus pernio on the face of a patient with sarcoidosis.
Ocular disease affects approximately one quarter of patients with systemic sarcoidosis and may involve any area of the eye. Uveitis is the most common ocular manifestation of sarcoidosis, affecting 25 percent of patients with the disorder, but sarcoidosis is responsible for only 5 to 7 percent of cases of uveitis overall.
Ocular disease involves the anterior segment of the eye in about 80 percent of patients and the posterior segment of the eye in about 20 percent of patients, usually in the form of chorioretinitis. The optic nerve may become involved in patients with neurosarcoidosis. Other ocular manifestations of sarcoidosis include cataracts, blindness, lacrimal gland swelling and inflammation, and retinal periphlebitis. The combination of keratoconjunctivitis and xerostomia, as in Sjögren's syndrome, has also occurred. Uveoparotid fever (also called Heerfordt's syndrome) is also known to affect patients with sarcoidosis. Symptoms include painless swelling of the parotid gland, fever and uveitis. Rarely, cranial nerve involvement (i.e., paralysis of the facial nerve) has been associated with this syndrome.
Sarcoid granulomas commonly infiltrate the lymph nodes and salivary glands. When hilar nodes are included, the incidence of lymphadenopathy in patients with sarcoidosis probably approaches 90 percent; the incidence of peripheral lymphadenopathy is about 27 percent. Lymphadenopathy can be associated with both acute and chronic disease and is usually painless. A peripheral lymph node may at times be easily accessible for biopsy, as these lymph nodes may remain enlarged for several years. Sarcoidosis affects the parotid glands in approximately 6 percent of patients, causing a complaint of “dry mouth.”
Renal involvement is clinically diagnosed in only 1 percent of patients with sarcoidosis but is found in up to 20 percent of patients in renal biopsy and necropsy studies.7 Three types of renal involvement have been described, the most common being nephrocalcinosis and/or kidney stones. Others include direct granulomatous involvement and glomerulonephritis. Patients with sarcoidosis have an altered ability to metabolize vitamin D, which often leads to enhanced absorption of intestinal calcium. Even in the absence of hypercalcemia there is often persistent absorptive hypercalciuria. Calcium nephropathy associated with nephrocalcinosis and calcium-containing kidney stones may lead to renal failure. Rarely, extensive granulomatous involvement may produce marked interstitial nephritis and parenchymal destruction with modest proteinuria and sterile pyuria. A patient with glomerulonephritis usually responds well to glucocorticoid therapy.
Fever is not a feature of sarcoidosis, except in certain circumstances. It almost always accompanies erythema nodosum, polyarthralgia and hilar adenopathy, and is usually self-limiting. It frequently occurs in black patients with significant hepatic involvement and also in patients with extensive retroperitoneal adenopathy.8
Hepatic involvement in patients with sarcoidosis represents a continuum from the presence of mostly asymptomatic granulomas to cases in which hepatic involvement represents a prominent part of the overall clinical picture. Approximately 20 percent of patients have hepatic involvement at some time during the course of the disease. Up to 40 percent of these patients have abnormal liver function tests, most commonly mild elevation of alkaline phosphatase levels. Alanine transaminase, aspartate transaminase and bilirubin levels are usually normal. About 10 percent of patients with sarcoidosis have hepatomegaly. Hepatic involvement can take several forms, including hepatomegaly (generally accompanied by splenomegaly and abnormal liver function tests), chronic cholestasis, and portal hypertension and cirrhosis, although the latter two are rare. Splenic involvement in patients with sarcoidosis is less common, occurring in 10 to 13 percent of patients.
Liver biopsy may be of value in establishing a diagnosis of sarcoidosis. However, care must be taken in the interpretation of hepatic granulomas since several other causes of non-caseating granulomas have been reported. Diseases such as tuberculosis and fungal infections must be ruled out before initiating corticosteroid therapy. The characteristic histologic feature of hepatic sarcoidosis is the presence of granulomas, frequently located in portal tracts. Chronic portal tract inflammation, fibrosis and even cirrhosis may accompany this finding. Liver biopsy is a valuable procedure in establishing a diagnosis of sarcoidosis, but positive findings must be correlated with evidence of sarcoid disease elsewhere in the body.
Cardiac sarcoidosis occurs in 3 to 5 percent of patients. It is very difficult to recognize clinically and is more often found at autopsy. A patient with sarcoidosis is considered to have cardiac sarcoidosis if bundle branch block, tachyarrhythmia, bradyarrhythmia, congestive heart failure, pericarditis or cardiomyopathy develop.9 Sudden death resulting from rhythm disturbances without prior symptoms occurs in up to 25 percent of patients with cardiac sarcoidosis, especially in patients under 40 years of age. Other manifestations include the formation of ventricular aneurysm, angina and papillary muscle dysfunction.
Bone sarcoidosis occurs in 3 to 4 percent of patients. It most frequently affects the hands and feet, and is associated with soft tissue swelling, joint stiffness and pain. Occasionally the gait may become affected. The patient with bone sarcoidosis usually complains of polyarthralgia, which most frequently affects the small joints of the hands and feet. Radiographs of the affected areas may show cystic or lytic lesions. Bone involvement reflects chronic irreversible progressive sarcoidosis and is common in conjunction with skin lesions, particularly lupus pernio. Chronic sarcoid arthritis appears to be more common in blacks and clinically may appear as a rheumatoid-like arthritis.
Neurosarcoidosis occurs in less than 5 percent of patients with sarcoidosis and is a serious complication of the disease. Early in the course of the disease, neurosarcoidosis may affect the central nervous system. Years later, the peripheral nervous system may become affected. This complication is associated with substantial mortality. Several possible manifestations of neurosarcoidosis have been reported, including involvement of cranial nerves (especially the facial and optic nerves), encephalopathy, seizures, cerebellar signs (such as ataxia or tremor), hearing loss, peripheral neuropathy and space-occupying masses.
Magnetic resonance imaging (MRI) and lumbar puncture, with findings of elevated protein levels, decreased glucose levels and pleocytosis, are helpful in making a diagnosis.
Almost any area of the body can be affected by sarcoidosis. Bone marrow granulomas, gastric granulomas, gonadal lesions, spinal cord lesions and pleural granulomas have been reported but only rarely. Hoarseness and epistaxis may occur in the upper respiratory tract as a result of excessive dryness and inflammation. Pituitary dysfunction leading to diabetes insipidus has also been reported.
Diagnosis
The diagnosis of sarcoidosis requires three components: the presence of clinical and radiographic findings consistent with a diagnosis of sarcoidosis; non-caseating (non-necrotizing) granulomas found on biopsies obtained from one or more sites; and exclusion of other granuloma-forming diseases using appropriate histochemical, microbiologic and serologic tests. Granulomas consist of a central follicular area with macrophages, epithelioid cells and multinucleated giant cells, surrounded by lymphocytes, fibroblasts and monocytes at the periphery2 (Figure 8). A thorough history and physical examination are imperative and should include queries regarding occupational and environmental exposure to potential pulmonary pathogens.
FIGURE 8.
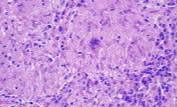
High-power view of non-caseating granuloma, which histologically supports the diagnosis of sarcoidosis.
As a rule, biopsies are obtained from the most obvious or the most frequent sites of clinical activity and should also be sent for cultures and special staining. Because pulmonary involvement is frequently a feature of sarcoidosis, fiberoptic transbronchial biopsies are often obtained. A transbronchial biopsy will demonstrate sarcoidosis in 60 percent of patients with stage 1 disease and more than 80 percent of patients with stage 2 or stage 3 disease. In the past, bronchoalveolar lavage has been performed at the time of bronchoscopy as a means of evaluating the “activity” of the disease, but this procedure is currently considered to be mainly of research interest. If results of bronchoscopic biopsies do not yield a diagnosis, biopsies of the hilar lymph nodes, taken during mediastinoscopy, demonstrate sarcoidosis in over 95 percent of patients.
Other useful sites of biopsy in a patient suspected of having sarcoidosis include the skin, the peripheral lymph nodes and the salivary glands. The skin should be closely examined for the presence of suspicious papules, nodules, plaques or scars. The skin is very accessible and skin biopsies offer a high yield for the diagnosis of sarcoidosis. Biopsies of palpable peripheral lymph nodes should be obtained when feasible. The minor salivary glands of the lower lip are another site for biopsy, yielding a diagnosis in 55 to 60 percent of patients. Biopsies of the liver demonstrate sarcoidosis in 60 to 75 percent of patients. However, liver biopsy has associated morbidity and should be performed only in patients in whom the other modalities have failed to yield a diagnosis. Bone marrow is not usually a high-yield tissue and should not routinely be biopsied.
Serum angiotensin converting enzyme is elevated in 50 to 80 percent of patients with sarcoidosis. The enzyme is secreted by epithelioid cells at the periphery of the granulomas, and the level is usually elevated in patients with “active” sarcoidosis.2 Because the enzyme is also present in pulmonary capillary endothelium, the level of serum angiotensin converting enzyme is higher in patients with intrathoracic involvement. Levels of serum angiotensin converting enzyme are most helpful to the clinician in following the course and “activity” of the disease and in evaluating treatment.8 Elevated levels of serum angiotensin converting enzyme alone do not confirm the presence of sarcoidosis, as these levels may be elevated in patients with such varying conditions as miliary tuberculosis, silicosis, asbestosis, biliary cirrhosis, leprosy, histoplasmosis, hepatitis, lymphoma, berylliosis, diabetic retinopathy and hyperthyroidism. If clinical suspicion of sarcoidosis exists and nonspecific granulomas are present but their atypical presentation makes the diagnosis difficult, determination of an elevated serum angiotensin converting enzyme level may help to confirm the diagnosis.
Anergy skin testing usually demonstrates cutaneous hyporeactivity to common antigens, (e.g., mumps virus or Candida species). A lower ratio of helper T cells to suppressor-cytotoxic T cells in the peripheral blood may explain the cutaneous anergy, which is found in up to one half of patients with sarcoidosis2 and reflects depression of delayed-type hypersensitivity. The purified protein derivative skin test for tuberculosis may be negative in up to 60 percent of these patients.
In addition to chest radiography, other tests and procedures that are useful as part of the work-up in patients with suspected sarcoidosis include pulmonary function testing, serum calcium determination, electrocardiography and slit lamp testing. While some patients with sarcoidosis have test results revealing obstructive changes, most results demonstrate restrictive lung disease and a decrease in carbon monoxide diffusing capacity.Vital capacity and pulmonary compliance may also be diminished. It is important to remember that results of pulmonary function testing may correlate poorly with radiographic findings. Hypercalcemia and hypercalciuria are common in patients with sarcoidosis and, if present, should be treated. If left untreated, nephrocalcinosis, renal failure and death may result. Patients with sarcoidosis may have an increased absorption of calcium from the gastrointestinal tract. An electrocardiogram should be performed on all patients with sarcoidosis to determine the presence of conduction abnormalities. If abnormalities are found or cardiac symptoms are present, 24-hour ambulatory monitoring, echocardiography and stress testing may be performed. Patients should undergo slit lamp examination at the time of initial diagnosis and annually to assess for ocular changes. Renal function tests and determination of liver enzyme, total serum protein and gamma globulin levels are also useful in evaluating for the presence of extrathoracic disease.
Several other tests have been used to help diagnose sarcoidosis, but they are considered very nonspecific. They include determinations of serum lysozyme levels, which may be elevated in patients with sarcoidosis and many other inflammatory disorders and are therefore not very useful; the Kveim-Siltzbach skin test (mentioned for historic reasons since it is now used in research only because of the inherent danger of using an injection of antigen from the sarcoid tissue of another patient); radioisotope scanning with gallium-67, which is a fairly good index of active alveolitis since sarcoid granulomas will actively take up gallium (this test is nonspecific and can be costly but may be useful in patients with “normal” findings on chest radiographs); and, finally, high-resolution computed tomography of the chest, which may be better than MRI in evaluating for parenchymal disease.
Treatment
The goals of treatment for sarcoidosis include resolving inflammatory lesions that are interfering with organ function, preventing pulmonary fibrosis and diminishing symptoms.3 If the patient presents with stage 1 or stage 2 disease with normal pulmonary function tests and no life-threatening signs or symptoms, observation is all that is necessary, as sarcoidosis is usually a self-limited disease and does not require specific therapy. Treatment is indicated if the patient has systemic symptoms or if deterioration in lung function is present at any stage, or if the patient presents with or progresses to stage 3 disease.
Corticosteroids continue to be the mainstay of therapy, although they have not been proved to prolong life. Several different protocols exist. To induce disease regression, treatment with prednisone may be started at a dosage of 40 to 60 mg per day given in divided doses for six to eight weeks, then tapered to a dosage of 15 to 20 mg per day over four to six months. A dosage of 40 to 60 mg of prednisone every other day has also been used for initial treatment, with excellent results.3,8 A patient may then be maintained on a dosage of 5 to 10 mg per day to suppress disease activity for up to one year. Patients should receive treatment if they have the following forms of sarcoidosis: hypercalcemia and hypercalciuria, disfiguring skin lesions, ocular sarcoidosis (this should be treated with topical and/or systemic steroids), cardiac sarcoidosis, neurologic sarcoidosis and other organ involvement that is determined to be clinically severe.8
Relapse occurs in 25 to 40 percent of patients with sarcoidosis within two to three months after discontinuing corticosteroid therapy. If this occurs, clinical examination and laboratory testing should be repeated. Some experts utilize “pulse therapy”with intravenous methylprednisolone at a dosage of 3 g per day for three days during acute exacerbations.3
Inhaled steroids have been used in patients with sarcoidosis for relief of symptoms, but it has not been proved that this therapy reduces disease progression. Inhaled and oral bronchodilators, supplemental oxygen and synthetic “liquid” tears have also been used to reduce symptoms. Topical ophthalmic steroids have been used to reduce ocular manifestations of sarcoidosis. If symptoms of erythema nodosum and arthritis are present in patients with stage 2 disease, a nonsteroidal anti-inflammatory drug such as indomethacin (Indocin), in a dosage of 25 mg three times daily, may be used.8
Newer therapies have been reported. Hydroxychloroquine (Plaquenil), given in a dosage of 200 mg every other day for nine months, may be useful in the treatment of cutaneous sarcoidosis but can permanently damage the eyes; consequently, ocular examinations must be performed frequently. Hydroxychloroquine has also been found to be helpful in the management of hypercalcemia.1 Methotrexate (Rheumatrex), in a low dosage of 7.5 to 15 mg once per week, has been shown to be of benefit in the treatment of refractory sarcoidosis, especially musculoskeletal and cutaneous forms.1 Other treatments are available, but few controlled trials have been performed: chlorambucil (Leukeran), cyclophosphamide (Cytoxan) and azathioprine (Imuran). Rarely, lung transplantation has been performed in patients with severe, refractory disease, with varying results. Several patients had a recurrence of granulomatous disease in the transplanted lung.2,10
Prognosis
The prognosis of patients with sarcoidosis is generally good, with spontaneous resolution of disease being the “rule.” Mortality rates vary from 5 to 8 percent.2 The prognosis is usually better in women, in patients with less severe pulmonary staging at the time of diagnosis, in patients who do not exhibit anergy and in patients with normal globulin levels. Proper management of sarcoidosis can clearly improve the quality of a patient's life.
