A 48-year-old woman presented with a lower extremity rash (Figures 1 and 2) that had persisted for three months. The rash appeared in recurrent crops in different areas; each lasted three to four weeks. The rash was accompanied by a stinging sensation and residual hyperpigmentation. She denied having fever, arthralgia, abdominal pain, diarrhea, or cough. She was not taking medications and had no history of intravenous drug use, hepatitis, transfusion, recent travel, exposure to chemicals, or allergies.
Figure 1.
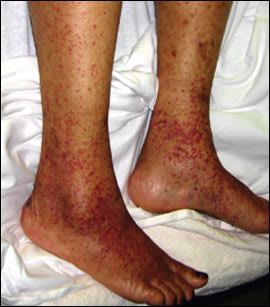
Figure 2.
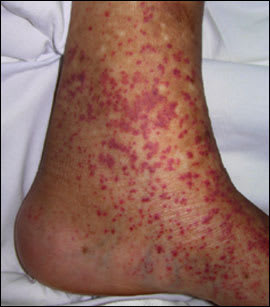
Examination revealed purpuric lesions on the legs and ankles ranging in size from pinpoint to 0.5 cm. The rash was palpable and nonblanching. Punch biopsy of the affected skin showed leukocytoclastic vasculitis. No immunoglobulin A (IgA) deposits were noted on immunofluorescence.
Laboratory testing revealed blood counts within normal limits (including a normal platelet count of 300 × 103 per μL [300 × 109 per L]), no electrolyte abnormalities, an erythrocyte sedimentation rate of 40 mm per hour, and normal renal function. Test results were negative for rheumatoid factor, antinuclear antibodies, cryoglobulins, cytoplasmic-type antineutrophil cytoplasmic autoantibody, anticardiolipin antibody, lupus anticoagulant, and hepatitis panel. Complement levels and serum immunoglobulin levels were normal, and peripheral blood smear and urinalysis results were unremarkable.
Question
Which one of the following is the most likely diagnosis?
A. Essential mixed cryoglobulinemia.
B. Henoch-Schönlein purpura.
C. Idiopathic hypersensitivity vasculitis.
D. Schamberg disease.
E. Thrombotic thrombocytopenic purpura.
Discussion
The answer is C: idiopathic hypersensitivity vasculitis. Idiopathic hypersensitivity vasculitis is diagnosed only after other causes of leukocytoclastic vasculitis are ruled out with patient history and laboratory testing. The cause of hypersensitivity vasculitis remains unknown in as many as 50 percent of patients.1 Careful patient evaluation is needed to rule out systemic involvement and to define its type and severity. Laboratory screening tests are useful in confirming the diagnosis and detecting any associated underlying disease. A punch biopsy of the affected skin can confirm the presence of vasculitis and provide clues to the underlying process.2
Essential mixed cryoglobulinemia may occur in patients with chronic hepatitis C virus infection, other infections, plasma cell or lymphoid neoplasms, or inflammatory diseases. Patients have neuropathy, arthritis, and renal involvement with detectable cryoglobulins and abnormal complement levels. The condition rarely occurs as an idiopathic process.3
Henoch-Schönlein purpura involves IgA deposits in blood vessel walls. Typical features include arthritis or arthralgia, abdominal pain, and renal disease.4
Schamberg disease (progressive pigmentary dermatosis) is an asymptomatic, non-palpable, brownish, macular eruption of the skin that usually occurs on the lower extremities and ankle area. It is caused by capillary leakage of blood, which, after subsequent breakdown, leaves hemosiderin staining and the characteristic cayenne pepper spots.5
Patients with thrombotic thrombocytopenic purpura have an abnormal platelet count, schistocytes on a peripheral smear, and abnormal renal function. They also may have altered neurologic status and a prodrome of bloody diarrhea or fever.6
Selected Differential Diagnosis of a Stinging Rash of Indeterminate Origin
| Condition | Characteristics |
|---|---|
| Essential mixed cryoglobulinemia | Palpable purpura, LCV, neuropathy, arthritis, renal involvement, hypocomplementemia, cryoglobulins |
| Henoch-Schönlein purpura | Palpable purpura, LCV with immunoglobulin A deposits in blood vessel walls, arthritis or arthralgia, abdominal pain, renal involvement |
| Idiopathic hypersensitivity vasculitis | Palpable purpura, LCV, elevated erythrocyte sedimentation rate, no offending medications or underlying cause detected despite extensive work-up |
| Schamberg disease | Asymptomatic, nonpalpable, brownish, macular eruption |
| Thrombotic thrombocytopenic purpura | Thrombocytopenia, schistocytes on peripheral smear, renal insufficiency, may cause neurologic symptoms |
LCV = leukocytoclastic vasculitis.