The thalassemias are a group of inherited hematologic disorders caused by defects in the synthesis of one or more of the hemoglobin chains. Alpha thalassemia is caused by reduced or absent synthesis of alpha globin chains, and beta thalassemia is caused by reduced or absent synthesis of beta globin chains. Imbalances of globin chains cause hemolysis and impair erythropoiesis. Silent carriers of alpha thalassemia and persons with alpha or beta thalassemia trait are asymptomatic and require no treatment. Alpha thalassemia intermedia, or hemoglobin H disease, causes hemolytic anemia. Alpha thalassemia major with hemoglobin Bart's usually results in fatal hydrops fetalis. Beta thalassemia major causes hemolytic anemia, poor growth, and skeletal abnormalities during infancy. Affected children will require regular lifelong blood transfusions. Beta thalassemia intermedia is less severe than beta thalassemia major and may require episodic blood transfusions. Transfusion-dependent patients will develop iron overload and require chelation therapy to remove the excess iron. Bone marrow transplants can be curative for some children with beta thalassemia major. Persons with thalassemia should be referred for preconception genetic counseling, and persons with alpha thalassemia trait should consider chorionic villus sampling to diagnose infants with hemoglobin Bart's, which increases the risk of toxemia and postpartum bleeding. Persons with the thalassemia trait have a normal life expectancy. Persons with beta thalassemia major often die from cardiac complications of iron overload by 30 years of age.
The thalassemias (named from the Greek word for sea, thalassa1) are a group of inherited autosomal recessive hematologic disorders2 that cause hemolytic anemia because of the decreased or absent synthesis of a globin chain. Imbalances of globin chains cause hemolysis and impair erythropoiesis. Family physicians need to know how to diagnose thalassemias, how to distinguish them from other causes of a microcytic anemia, and the treatment options for severe forms of thalassemia.
Epidemiology
Approximately 5 percent of the world's population has a globin variant, but only 1.7 percent has alpha or beta thalassemia trait.2 Thalassemia affects men and women equally and occurs in approximately 4.4 of every 10,000 live births. Alpha thalassemia occurs most often in persons of African and Southeast Asian descent, and beta thalassemia is most common in persons of Mediterranean, African, and Southeast Asian descent. Thalassemia trait affects 5 to 30 percent of persons in these ethnic groups.2
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Persons with anemia from thalassemia trait should not take iron supplements unless they have coexistent iron deficiency. | C | 2, 6 |
| Persons with beta thalassemia major require periodic lifelong blood transfusions to maintain hemoglobin levels higher than 9.5 g per dL (95 g per L) and sustain normal growth. | B | 2, 15 |
| Persons with beta thalassemia major require chelation therapy for iron overload. | A | 16 |
| Persons at risk of having a child with thalassemia should be offered preconception genetic counseling. | C | 20, 21 |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.xml.
Pathophysiology
Hemoglobin consists of an iron-containing heme ring and four globin chains: two alpha and two nonalpha. The composition of the four globin chains determines the hemoglobin type. Fetal hemoglobin (HbF) has two alpha and two gamma chains (alpha2 gamma2). Adult hemoglobin A (HbA) has two alpha and two beta chains (alpha2 beta2), whereas hemoglobin A2 (HbA2) has two alpha and two delta chains (alpha2 delta2). At birth, HbF accounts for approximately 80 percent of hemoglobin and HbA accounts for 20 percent.3 The transition from gamma globin synthesis (HbF) to beta globin synthesis (HbA) begins before birth. By approximately six months of age, healthy infants will have transitioned to mostly HbA, a small amount of HbA2, and negligible HbF. Figure 1 shows normal and abnormal hemoglobins.
Figure 1.
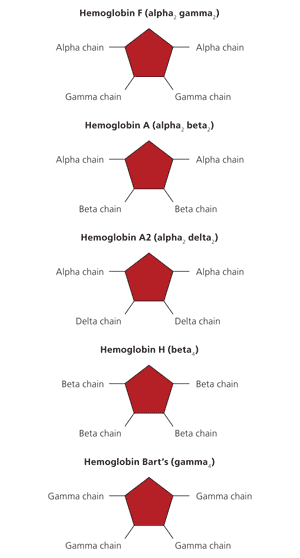
Normal (hemoglobin F, A, and A2) and abnormal (hemoglobin H and Bart's) hemoglobins. Hemoglobin consists of an iron-containing heme ring and four globin chains: two alpha and two nonalpha. The composition of the four globin chains determines the hemoglobin type.
ALPHA THALASSEMIA
Alpha thalassemia is the result of deficient or absent synthesis of alpha globin chains, leading to excess beta globin chains. Alpha globin chain production is controlled by two genes on each chromosome 16 (Table 14,5). Deficient production is usually caused by a deletion of one or more of these genes. A single gene deletion results in alpha thalassemia silent carrier status, which is asymptomatic with normal hematologic findings. The two-gene deletion causes alpha thalassemia trait (minor) with microcytosis and usually no anemia. The three-gene deletion results in significant production of hemoglobin H (HbH), which has four beta chains (beta4). Alpha thalassemia intermedia, or HbH disease, causes microcytic anemia, hemolysis, and splenomegaly. The four-gene deletion results in significant production of hemoglobin Bart's (Hb Bart's), which has four gamma chains (gamma4). Alpha thalassemia major with Hb Bart's usually results in fatal hydrops fetalis.
Table 1. Prototypical Forms of Alpha Thalassemia
| Variant | Chromosome 16 | Signs and symptoms |
|---|---|---|
| Alpha thalassemia silent carrier | One of four gene deletions | Asymptomatic |
| Alpha thalassemia trait | Two of four gene deletions | Asymptomatic |
| Hemoglobin Constant Spring | Reduced output of alpha globin | Silent or mildly symptomatic |
| Alpha thalassemia intermedia with significant hemoglobin H (hemoglobin H disease) | Three of four gene deletions | Moderate to severe hemolytic anemia, modest degree of ineffective erythropoiesis, splenomegaly, variable bone changes4 |
| Alpha thalassemia major with significant hemoglobin Bart's | Four of four gene deletions | Causes nonimmune hydrops fetalis, usually fatal5 |
BETA THALASSEMIA
Beta thalassemia is the result of deficient or absent synthesis of beta globin chains, leading to excess alpha chains. Beta globin synthesis is controlled by one gene on each chromosome 11. Beta thalassemia occurs from any of more than 200 point mutations and (rarely) deletions of the two genes. Beta globin chain production can range from near normal to completely absent, leading to varying degrees of excess alpha globin to beta globin chain production. The one gene defect, beta thalassemia trait (minor), is asymptomatic and results in microcytosis and mild anemia. If the synthesis from both genes is severely reduced or absent, the person has beta thalassemia major, also known as Cooley anemia. Persons with beta thalassemia major are almost never symptomatic at birth because of the presence of HbF, but symptoms begin to develop by six months of age. If the synthesis of beta chains is less severely reduced, the person has beta thalassemia intermedia. These persons experience symptoms that are less severe and do not require lifelong transfusions to survive past 20 years of age (Table 2).6
Table 2. Prototypical Forms of Beta Thalassemia
| Variant | Chromosome 11 | Signs and Symptoms |
|---|---|---|
| Beta thalassemia trait | One gene defect | Asymptomatic |
| Beta thalassemia intermedia | Two genes defective (mild to moderate decrease in beta globin synthesis) | Variable degrees of severity of symptoms of thalassemia major |
| Beta thalassemia major | Two genes defective (severe decrease in beta globin synthesis) | Abdominal swelling, growth retardation, irritability, jaundice, pallor, skeletal abnormalities, splenomegaly; requires lifelong blood transfusions6 |
Information from reference 6.
HEMOGLOBINOPATHIES WITH THALASSEMIA
A hemoglobinopathy is a genetic defect that results in an abnormal structure of a globin chain. A thalassemia results in an abnormally low quantity of a globin chain. Rarely, persons will have coexisting hemoglobinopathy and thalassemia (Online Table A).
Diagnosis
Most persons with thalassemia trait are found incidentally when their complete blood count shows a mild microcytic anemia. Microcytic anemia can be caused by iron deficiency, thalassemia, lead poisoning, sideroblastic anemia, or anemia of chronic disease. The mean corpuscular volume (MCV), red blood cell distribution width (RDW), and the patient's history can exclude some of these etiologies. The MCV is usually less than 75 fl with thalassemia and rarely less than 80 fl in iron deficiency until the hematocrit is less than 30 percent. For children, the Mentzer index (MCV/red blood cell count) can help distinguish between iron deficiency and thalassemia. In iron deficiency, the ratio is usually greater than 13, whereas thalassemia yields values less than 13. A ratio of 13 would be considered uncertain.7
The RDW may assist in differentiating iron deficiency and sideroblastic anemia from thalassemia (Table 3). The RDW will be elevated in more than 90 percent of persons with iron deficiency, but in only 50 percent of persons with thalassemia.8 The RDW is usually elevated in sideroblastic anemia. Therefore, although a microcytic anemia with a normal RDW will almost always be because of thalassemia, persons with an elevated RDW will require additional testing (Figure 2).9
Table 3. Hematologic Indices of Iron Deficiency and Alpha and Beta Thalassemia
| Test | Iron deficiency | Beta thalassemia | Alpha thalassemia |
|---|---|---|---|
| MCV (abnormal if < 80 fl in adults; < 70 fl in children six months to six years of age; and < 76 fl in children seven to 12 years of age) | Low | Low | Low |
| Red blood cell distribution width | High | Normal; occasionally high | Normal |
| Ferritin | Low | Normal | Normal |
| Mentzer index for children (MCV/red blood cell count) | > 13 | < 13 | < 13 |
| Hb electrophoresis | Normal (may have reduced HbA2) | Increased HbA2, reduced HbA, and probably increased HbF | Adults: normal Newborns: may have HbH or Hb Bart's |
Hb = hemoglobin; HbF = fetal hemoglobin; MCV = mean corpuscular volume.
Figure 2. Use of RDW Values in the Diagnosis of Thalassemia
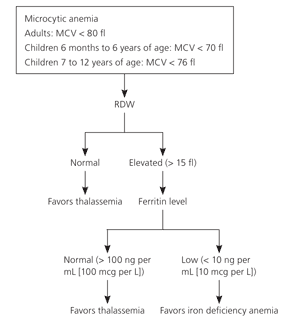
Algorithm of the use of RDW values to assist in diagnosing thalassemia. (MCV = mean corpuscular value; RDW = red blood cell distribution width.)
Information from reference 9.
Supplemental tests include serum ferritin, the peripheral smear, hemoglobin electrophoresis, serum lead level, and rarely bone marrow aspirate. Serum ferritin is the best test to screen for iron deficiency anemia.10 In the absence of inflammation, a normal ferritin level generally excludes iron deficiency. Serum iron, total iron-binding capacity, and transferrin saturation are rarely needed. Sideroblastic anemia can be excluded with an examination of the peripheral smear or the bone marrow aspirate. A normal serum lead level excludes lead poisoning. Anemia of chronic disease is most often a normocytic normochromic mild anemia. If thalassemia is still suspected, a hemoglobin electrophoresis may help diagnose the condition.
The hemoglobin electrophoresis with beta thalassemia trait usually has reduced or absent HbA, elevated levels of HbA2, and increased HbF.2 However, a normal concentration of HbA2 does not rule out beta thalassemia trait, especially if there was coexistent iron deficiency, which can lower HbA2 levels into the normal range. In the newborn period, if the electrophoresis shows Hb Bart's or HbH, the infant has alpha thalassemia. The hemoglobin electrophoresis is usually normal in adults with alpha thalassemia trait.
Persons with beta thalassemia major are diagnosed during infancy. Pallor, irritability, growth retardation, abdominal swelling, and jaundice appear during the second six months of life.6
Persons with a microcytic anemia but milder symptoms that start later in life have beta thalassemia intermedia.
Complications
The complications that occur with beta thalassemia major or intermedia are related to overstimulation of the bone marrow, ineffective erythropoiesis, and iron overload from regular blood transfusions. Untreated infants have poor growth, skeletal abnormalities, and jaundice. Alpha thalassemia intermedia, or HbH disease, causes hemolysis and severe anemia. Alpha thalassemia major with Hb Bart's causes nonimmune hydrops fetalis in utero, which is almost always fatal.
With multiple blood transfusions and continued absorption of intestinal iron, iron overload develops. Iron is deposited in visceral organs (mainly the heart, liver, and endocrine glands), and most patient deaths are caused by cardiac complications.11 Endocrinopathies, particularly hypogonadism and diabetes mellitus, may occur in adolescents and adults.2
Splenomegaly invariably develops in the symptomatic thalassemias. Splenomegaly can worsen the anemia and occasionally cause neutropenia and thrombocytopenia.
Thromboembolic events, venous and arterial, are not uncommon. Persons with beta thalassemia major or intermedia may develop a chronic hypercoagulable state,12 especially after splenectomy.13
Osteopenia and osteoporosis are being found more often as persons with thalassemia major live longer. One study found osteoporosis in 51 percent of persons older than 12 years with thalassemia major.14
General Management Issues
Persons with thalassemia trait require no treatment or long-term monitoring. They usually do not have iron deficiency, so iron supplements will not improve their anemia. Accordingly, iron therapy should only be administered if iron deficiency occurs.2,6
BLOOD TRANSFUSIONS
Persons with beta thalassemia major require periodic and lifelong blood transfusions to maintain a hemoglobin level higher than 9.5 g per dL (95 g per L) and sustain normal growth.2,15 The need for blood transfusions may begin as early as six months of age. For persons with beta thalassemia intermedia, the decision to transfuse is a more subjective clinical assessment. Transfusion requirements are episodic and become necessary when the person's hemoglobin is inadequate for a normal life or when the anemia impairs growth and development.
Alpha thalassemia intermedia, or HbH disease, causes mild to moderate hemolysis. Transfusions will occasionally be necessary depending upon the severity of the clinical condition.
CHELATION
Transfusion-dependent patients develop iron overload because they have no physiologic process to remove excess iron from multiple transfusions. Therefore, they require treatment with an iron chelator starting between five and eight years of age.16 Deferoxamine (Desferal), subcutaneously or intravenously, has been the treatment of choice. Although this therapy is relatively nontoxic, it is cumbersome and expensive. The U.S. Food and Drug Administration recently approved oral deferasirox (Exjade) as an alternative treatment.17 Adverse effects of deferasirox were transient and gastrointestinal in nature, and no cases of agranulocytosis were reported.
BONE MARROW TRANSPLANT
Bone marrow transplantation in childhood is the only curative therapy for beta thalassemia major. Hematopoietic stem cell transplantation generally results in an excellent outcome in low-risk persons, defined as those with no hepatomegaly, no portal fibrosis on liver biopsy, and regular chelation therapy, or at most, two of these abnormalities.2
Management of Specific Conditions
HYPERSPLENISM
If hypersplenism causes a marked increase in transfusion requirements, splenectomy may be needed. Surgery is usually delayed until at least four years of age because of the spleen's role in clearing bacteria and preventing sepsis. At least one month before surgery, patients should receive the pneumococcal polysaccharide vaccine. Children should also receive the pneumococcal conjugate vaccine series. Antibiotic prophylaxis with penicillin, 250 mg orally twice a day, is recommended for all persons during the first two years after surgery and for children younger than 16 years.18
ENDOCRINOPATHIES
Growth hormone therapy for beta thalassemia intermedia and major has had variable success and is not generally recommended. Hormone therapy is effective for hypogonadism.19
PREGNANCY
Preconception genetic counseling is strongly advised for all persons with thalassemia.20 Two parents, each with beta thalassemia trait, have a one in four chance of conceiving a child with beta thalassemia major and a three in four chance the child will have thalassemia trait or be normal (Online Figure A). Persons with alpha thalassemia trait have a more complex pattern of inheritance. Whether both defective genes are on the same or different chromosomes will alter the outcome (Online Figure B).
Chorionic villus sampling using polymerase chain reaction technology to detect point mutations or deletions can identify infants affected with beta thalassemia. Persons with alpha thalassemia trait should consider prenatal diagnosis because Hb Bart's increases the risk of toxemia and postpartum bleeding. Preimplantation genetic diagnosis is becoming available in conjunction with in vitro fertilization.21
CARDIAC
Serum ferritin has been used as a marker of iron storage to predict cardiac complications. Ferritin levels less than 2,500 ng per mL (2,500 mcg per L) are associated with improved survival.22 However, ferritin levels are unreliable when liver disease is present.23
HYPERCOAGULOPATHY
No randomized or observational studies regarding anticoagulation, antiplatelet therapy, or both have been reported for persons at greater risk of thromboembolic events. Therefore, no specific treatment can be recommended. Persons with a history of thrombosis may be treated with low-molecular-weight heparin. Anticoagulation therapy is warranted before surgery and during pregnancy for at-risk persons. Alternatives to estrogen-containing contraception should be offered to women of reproductive age.
PSYCHOSOCIAL
Beta thalassemia major or intermedia is a chronic disease with a significant impact on the patient and the patient's family and offspring. Education about the genetics of the disease, prenatal diagnosis options, and psychological therapy to help manage the complications is appropriate. However, neither the type of education nor the duration of therapy can be specified based on current evidence.24
VITAMIN DEFICIENCIES
Folic acid deficiency has been reported in thalassemia major and intermedia as a result of increased erythropoiesis. Therefore, daily oral supplementation with 1 mg of folic acid is recommended for persons with evidence of folate deficiency.18
Because some complications seem to be related to cellular oxidative stress, treatment with antioxidants has been thought to be beneficial.18 However, no improvements in anemia or reductions in morbidity or mortality have been demonstrated. Vitamin C is not recommended except in transfusion-dependent patients with a proven deficiency.
Prognosis
Persons with thalassemia trait have a normal life expectancy. Persons with beta thalassemia major live an average of 17 years and usually die by 30 years of age. Most deaths are caused by the cardiac complications of iron overload.11
