Proteinuria is common in children and may represent a benign condition or a serious underlying renal disease or systemic disorder. Proteinuria may occur secondary to glomerular or tubular dysfunction. Although a 24-hour urine protein excretion test is usually recommended, it may be impractical in children. A spot, first-morning urine test for protein/creatinine ratio can be useful in this situation. Proteinuria is usually benign, in the form of transient or orthostatic proteinuria. Persistent proteinuria may be associated with more serious renal diseases. Clinical features from the history, physical examination, and laboratory tests help determine the cause of proteinuria. Treatment should be directed at the underlying cause. Patients with active urinary sediments, persistent and gross hematuria, hypertension, hypocomplementemia, renal insufficiency with depressed glomerular filtration rate, or signs and symptoms suggestive of vasculitic disease may require a renal biopsy and referral to a pediatric nephrologist.
The presence of protein in urine is a common laboratory finding in children. Although proteinuria is usually benign, the condition can be a marker for a serious underlying renal disease or systemic disorder.1,2 When proteinuria coexists with hematuria, the likelihood of clinically significant renal disease is higher.2,3 The challenge for the primary care physician is to separate benign forms of proteinuria from those with clinical significance.
SORT: KEY RECOMMENDATIONS FOR PRACTICE

| Clinical recommendations | Evidence rating | References |
|---|---|---|
| A 24-hour urine protein excretion test is often difficult in children. Spot, first-morning urine testing for UPr/Cr is more useful and correlates well with the 24-hour urine collection. Spot, first- morning urine testing should be used in children if the urine dipstick test result is 1+ or more. | C | 2, 18–19 |
| When a child's UPr/Cr is greater than 0.2 (greater than 0.5 for children six to 24 months of age) or urinalysis results are abnormal, further investigation with history, physical examination, and blood work is recommended to rule out significant renal disease. | C | 2 |
| Referral to a pediatric nephrologist should be considered if a definitive diagnosis is required or a renal biopsy is considered. | C | 2 |
UPr/Cr = urine protein/creatinine ratio.
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.xml.
Epidemiology
Proteinuria is present at routine urine testing in up to 10 percent of school-aged children, although this decreases to 0.1 percent at repeated testing.4 A study including mass screening of school-aged children in Asia revealed similar findings.5–7 The prevalence increases with age, peaks during adolescence, and is higher in girls.8
Mechanism of Proteinuria
The glomerular barrier has three layers (the fenestrated endothelium, the basement membrane, and the podocytes), forming both a size-selective and electrostatic filter. 1,9,10 The electrostatic barrier consists of negatively charged sialoproteins and proteoglycans. Most proteins, such as immunoglobulins G and M, are too large (greater than 100 kDa) to pass through the glomerular barrier. Some have a charge or conformation that prevents them from traversing the filter. At least one half of the proteins in normal urine are Tamm-Horsfall proteins, which are localized to the thick ascending limbs of the loop of Henle.11 The remaining proteins are filtered plasma proteins of different molecular sizes, including mostly low-molecular-weight proteins (less than 40 kDa), such as transferrin, microglobulins, and intermediate-sized albumin.1,2,12,13 Most filtered proteins at the glomerulus are reabsorbed in the proximal tubule.
Slit diaphragms between podocytes have recently been discovered. These slit diaphragms contribute to the barrier effect. Mutations of the slit diaphragms can disrupt normal function and lead to proteinuria.9
Mechanisms of proteinuria can be categorized as glomerular, tubular, secretory, or overflow; glomerular and tubular are the primary mechanisms in children.10,12,14 Proteinuria may result from increased glomerular permeability due to damage to the integrity of the glomerular filter.10 Proteinuria can also occur when a reduced number of functioning nephrons leads to increased diffusion of protein across the remaining glomeruli. Tubular proteinuria occurs when there is an increased excretion of normally filtered low-molecular weight proteins due to impaired reabsorption by the proximal tubules.10,14 Secretory proteinuria results from oversecretion of certain proteins in the tubules, most notably the oversecretion of Tamm-Horsfall proteins in interstitial nephritis. Overflow proteinuria occurs when the plasma concentrations of low-molecular-weight proteins exceed the capacity of the tubules to reabsorb the filtered protein. Examples include hemoglobinuria in intravascular hemolysis and myoglobinuria in rhabdomyolysis.
Measurement of Proteinuria
OFFICE TESTING
The urine dipstick test uses the tetrabromophenol blue colorimetric method, which is the most widely used screening method. The intensity of color changes from yellow to blue and correlates with the amount of protein in the urine: trace (10 mg per dL), 1+ (30 mg per dL), 2+ (100 mg per dL), 3+ (300 mg per dL), and 4+ (1,000 mg per dL or greater).15 A reading of 1+ or more is considered abnormal. The dipstick test primarily detects albuminuria, with a specificity and sensitivity of more than 99 percent,16 but is not sensitive for other proteins. The dipstick test may yield false-positive results for proteinuria with alkaline urine (pH greater than 8), concentrated urine (specific gravity greater than 1.030), gross hematuria, pyuria, bacteriuria, prolonged immersion of reagent strip in the urine, placement of reagent strip directly in the urine stream, and presence of phenazopyridine or quaternary ammonium compounds in the urine.12 False-negative results may occur with acidic urine (pH less than 4.5), dilute urine (specific gravity less than 1.010), and presence of proteins other than albumin in the urine.12
The sulfosalicylic acid method, or turbidimetry, detects all forms of protein and is generally used as a supplementary test when the presence of a low-molecular-weight or other protein is suspected but not detected by the dipstick test. In the sulfosalicylic acid method, three drops of a sulfosalicylic acid 20% solution are added to 5 mL of urine. Depending on the amount of protein precipitated, various grades of turbidity from minimal (trace) to heavy flocculation (4+) are noted.2,3
QUANTITATIVE LABORATORY TESTING
The first-line test is 24-hour quantitative urine protein excretion. In children, the amount of urinary protein excretion varies by age and body size. The normal amount is less than 4 mg per m2 per hour or 100 mg per m2 per day.2 However, this quantitative measurement is not practical in children, particularly if they are incontinent.2 Also, it has an inherent time delay, is often difficult to obtain in an outpatient setting, and is subject to collection errors.
The single-void urine protein/creatinine ratio (UPr/Cr) calculated in milligrams of protein per milligrams of creatinine is a convenient method for estimating urine protein excretion without a 24-hour urine collection.2,17 Multiple studies have found that the 24-hour urine protein test correlates well with UPr/Cr.18–20 Multiplying UPr/Cr by 0.63 can give an estimate of the total amount of protein (g per m2 per day) in the urine. Tubular secretion of creatinine increases in the presence of a significant reduction in the glomerular filtration rate, and this might lead to an artificially low UPr/Cr.18 Nevertheless, the UPr/Cr is useful for following trends in proteinuria. A spot, first-morning urine sampling is optimal for determining UPr/Cr because it excludes any postural effect on the protein component.
Etiology
The etiology of proteinuria in children is diverse (Table 12,19,21,22 ), but a classification scheme based on the clinical timing and frequency of the problem can help narrow the differential diagnosis. The orthostatic and transient forms are benign and more common. Persistent proteinuria may be associated with underlying renal diseases and requires further investigation.
Table 1. Causes of Proteinuria in Children

| Transient (functional) proteinuria | ||
| Idiopathic | ||
| Related to medical condition (e.g., fever, seizure) | ||
| Unrelated to medical condition (e.g., exercise, stress, dehydration, cold exposure) | ||
| Orthostatic proteinuria | ||
| Persistent proteinuria | ||
| Glomerular | ||
| Adaptation (hyperfiltration) due to nephron loss (e.g., reflux nephropathy secondary to vesicoureteric reflux) | ||
| Alport syndrome | ||
| Collagen vascular disease or vasculitis: Henoch-Schönlein purpura, systemic lupus erythematosus | ||
| Diabetes mellitus | ||
| Glomerulopathy: minimal change glomerulopathy,* focal segmental glomerulosclerosis, mesangial proliferative glomerulonephritis, congenital nephrotic syndrome, immunoglobulin A nephropathy, membranoproliferative glomerulonephritis | ||
| Infection: group A beta-hemolytic streptococcus infection, viral infection (hepatitis B, hepatitis C, human immunodeficiency virus, infectious mononucleosis), other infection (malaria, syphilis) | ||
| Malignancies (lymphoma, solid tumors) | ||
| Toxin (mercury) | ||
| Tubulointerstitial | ||
| Acute tubular necrosis: aminoglycosides, cisplatin, amphotericin B, NSAIDs, radiocontrast media | ||
| Acute tubulointerstitial nephritis (NSAIDs, penicillin, cephalosporins, quinolones, sulfonamides, cimetidine [Tagamet], allopurinol [Zyloprim]) | ||
| Polycystic kidney disease | ||
| Proximal renal tubular acidosis: Fanconi syndrome (global proximal tubule dysfunction), cystinosis, Lowe syndrome, galactosemia, Wilson disease | ||
| Pyelonephritis | ||
| Toxins (lead, copper, mercury) | ||
NSAID = nonsteroidal anti-inflammatory drug.
*—Most common form of nephrotic syndrome.
TRANSIENT PROTEINURIA
Transient (functional) proteinuria is temporary and clears when the inciting factor remits or is removed. Transient proteinuria can occur with fever, exercise, stress, or cold exposure.17,23,24 It may also be caused by hemodynamic alterations in glomerular blood flow.
ORTHOSTATIC PROTEINURIA
Orthostatic proteinuria is not uncommon in children, particularly during adolescence. The diagnosis is suggested with normal protein excretion (i.e., negative dipstick test result or UPr/Cr of 0.2 or less) in a spot, first-morning urine sample after the child has been supine for the entire night, but increased protein excretion (i.e., positive dipstick test result or UPr/Cr greater than 0.2) at least four to six hours after the child has been upright.25 The cause of orthostatic proteinuria is not clear; however, the anatomic compression of the left renal vein has been suggested.26 Long-term studies with follow-up ranging from 20 to 50 years have demonstrated a benign course.27,28
PERSISTENT PROTEINURIA
Persistent proteinuria can be glomerular or tubulointerstitial in origin. In both categories, the causes can be primary, stemming intrinsically from the renal tissue; or secondary, mainly caused by systemic diseases. When proteinuria is associated with hematuria, renal dysfunction, and hypertension, significant renal disease may be present.2
Glomerular diseases are more common than tubulointerstitial diseases.2,29,30 Albumin and immunoglobulin G in the urine are the usual indicators for glomerular diseases. Glomerular diseases can have nephrotic and/or nephritic features, and making a distinction between these features can help narrow the differential diagnosis. Nephrotic syndrome is characterized by heavy proteinuria (greater than 1 g per m2 per day or UPr/Cr greater than 2.0), edema, hypoalbuminemia (less than 25 g per L), and hyperlipidemia.18,30–32 Nephritic features include hematuria; hypertension; oliguria; and active urinary sediments, such as red blood cells, white blood cells, and cellular casts.
Tubulointerstitial diseases are less common causes of proteinuria and usually involve low–molecular-weight proteins. Proteinuria associated with renal tubular disorders is generally mild. Tubular proteinuria rarely presents a diagnostic dilemma because the underlying disease is usually detected before the proteinuria.14,29
Interstitial nephritis includes a variety of pathologic processes involved in the progression of most renal diseases, and is a final common pathway for all forms of endstage renal disease.14
Diagnostic Evaluation
Proteinuria is often an incidental finding on urine dipstick testing or urinalysis. Asymptomatic children with proteinuria usually have the transient or orthostatic type. If a urine dipstick test shows trace amounts of protein, the test should be repeated with first-morning urine. If the first-morning test shows trace or negative amount of protein, a repeat first-morning test performed in one year should also be considered.2 In children with a urine dipstick test result of 1+ or greater, a spot, first-morning urine test for UPr/Cr and a urinalysis with microscopic examination should be performed. If the UPr/Cr is 0.2 or less (0.5 or less for children six to 24 months of age) and urinalysis results are normal, a diagnosis of transient or orthostatic proteinuria should be considered. A repeat first-morning dipstick test in one year should be considered. If the UPr/Cr is greater than 0.2 (greater than 0.5 for children six to 24 months of age), or if urinalysis results are abnormal (e.g., hematuria, leukocyturia, active urinary sediments), persistent proteinuria or proteinuria of clinical significance is more likely. A UPr/Cr greater than 2.0 is associated with nephrotic syndrome, and further evaluation with history, physical examination, and additional blood work is essential.2,29
History and Physical Examination
Because of the wide differential diagnosis of proteinuria in children, the symptoms and signs may vary. Common features of renal disease include growth failure, hypertension, and edema (i.e., periorbital, presacral, genital, or ankle). Associated deafness or visual impairment suggests hereditary nephritis. Table 2 lists clinical features that can provide clues to the underlying cause of persistent proteinuria.2,19,21,22
Table 2. Clinical Clues to the Cause of Persistent Proteinuria in Children
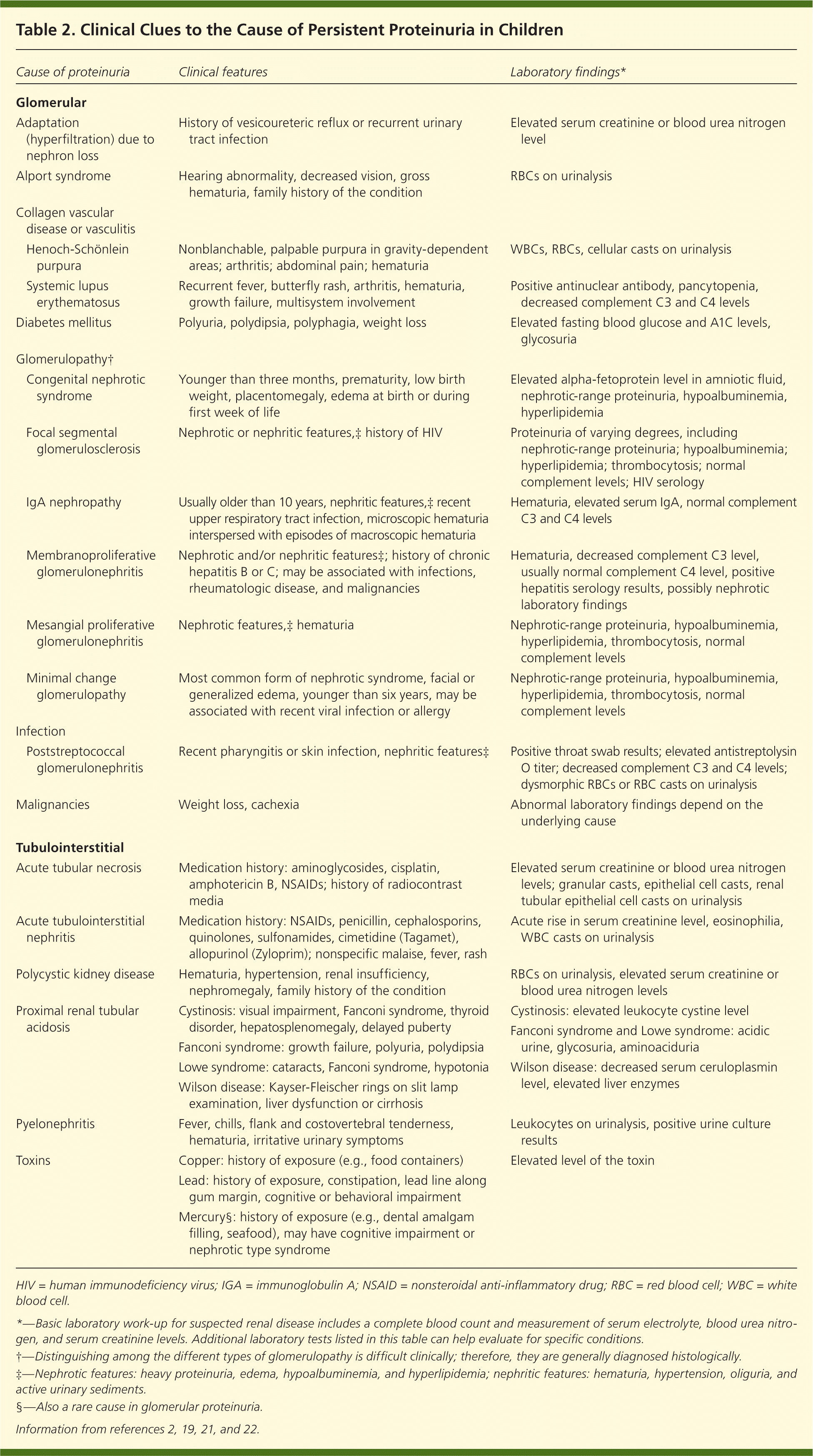
| Cause of proteinuria | Clinical features | Laboratory findings* | |
|---|---|---|---|
| Glomerular | |||
| Adaptation (hyperfiltration) due to nephron loss | History of vesicoureteric reflux or recurrent urinary tract infection | Elevated serum creatinine or blood urea nitrogen level | |
| Alport syndrome | Hearing abnormality, decreased vision, gross hematuria, family history of the condition | RBCs on urinalysis | |
| Collagen vascular disease or vasculitis | |||
| Henoch-Schönlein purpura | Nonblanchable, palpable purpura in gravity-dependent areas; arthritis; abdominal pain; hematuria | WBCs, RBCs, cellular casts on urinalysis | |
| Systemic lupus erythematosus | Recurrent fever, butterfly rash, arthritis, hematuria, growth failure, multisystem involvement | Positive antinuclear antibody, pancytopenia, decreased complement C3 and C4 levels | |
| Diabetes mellitus | Polyuria, polydipsia, polyphagia, weight loss | Elevated fasting blood glucose and A1C levels, glycosuria | |
| Glomerulopathy† | |||
| Congenital nephrotic syndrome | Younger than three months, prematurity, low birth weight, placentomegaly, edema at birth or during first week of life | Elevated alpha-fetoprotein level in amniotic fluid, nephrotic-range proteinuria, hypoalbuminemia, hyperlipidemia | |
| Focal segmental glomerulosclerosis | Nephrotic or nephritic features,‡ history of HIV | Proteinuria of varying degrees, including nephrotic-range proteinuria; hypoalbuminemia; hyperlipidemia; thrombocytosis; normal complement levels; HIV serology | |
| IgA nephropathy | Usually older than 10 years, nephritic features,‡ recent upper respiratory tract infection, microscopic hematuria interspersed with episodes of macroscopic hematuria | Hematuria, elevated serum IgA, normal complement C3 and C4 levels | |
| Membranoproliferative glomerulonephritis | Nephrotic and/or nephritic features‡; history of chronic hepatitis B or C; may be associated with infections, rheumatologic disease, and malignancies | Hematuria, decreased complement C3 level, usually normal complement C4 level, positive hepatitis serology results, possibly nephrotic laboratory findings | |
| Mesangial proliferative glomerulonephritis | Nephrotic features,‡ hematuria | Nephrotic-range proteinuria, hypoalbuminemia, hyperlipidemia, thrombocytosis, normal complement levels | |
| Minimal change glomerulopathy | Most common form of nephrotic syndrome, facial or generalized edema, younger than six years, may be associated with recent viral infection or allergy | Nephrotic-range proteinuria, hypoalbuminemia, hyperlipidemia, thrombocytosis, normal complement levels | |
| Infection | |||
| Poststreptococcal glomerulonephritis | Recent pharyngitis or skin infection, nephritic features‡ | Positive throat swab results; elevated antistreptolysin O titer; decreased complement C3 and C4 levels; dysmorphic RBCs or RBC casts on urinalysis | |
| Malignancies | Weight loss, cachexia | Abnormal laboratory findings depend on the underlying cause | |
| Tubulointerstitial | |||
| Acute tubular necrosis | Medication history: aminoglycosides, cisplatin, amphotericin B, NSAIDs; history of radiocontrast media | Elevated serum creatinine or blood urea nitrogen levels; granular casts, epithelial cell casts, renal tubular epithelial cell casts on urinalysis | |
| Acute tubulointerstitial nephritis | Medication history: NSAIDs, penicillin, cephalosporins, quinolones, sulfonamides, cimetidine (Tagamet), allopurinol (Zyloprim); nonspecific malaise, fever, rash | Acute rise in serum creatinine level, eosinophilia, WBC casts on urinalysis | |
| Polycystic kidney disease | Hematuria, hypertension, renal insufficiency, nephromegaly, family history of the condition | RBCs on urinalysis, elevated serum creatinine or blood urea nitrogen levels | |
| Proximalrenal tubular acidosis | Cystinosis: visual impairment, Fanconi syndrome, thyroid disorder, hepatosplenomegaly, delayed puberty Fanconi syndrome: growth failure, polyuria, polydipsia Lowe syndrome: cataracts, Fanconi syndrome, hypotonia Wilson disease: Kayser-Fleischer rings on slit lamp examination, liver dysfunction or cirrhosis | Cystinosis:elevated leukocyte cystine level Fanconi syndrome and Lowe syndrome: acidic urine, glycosuria, aminoaciduria Wilson disease: decreased serum ceruloplasmin level, elevated liver enzymes | |
| Pyelonephritis | Fever, chills, flank and costovertebral tenderness, hematuria, irritative urinary symptoms | Leukocytes on urinalysis, positive urine culture results | |
| Toxins | Copper: history of exposure (e.g., food containers) | Elevated level of the toxin | |
| Lead: history of exposure, constipation, lead line along gum margin, cognitive or behavioral impairment | |||
| Mercury§: history of exposure (e.g., dental amalgam filling, seafood), may have cognitive impairment or nephrotictype syndrome | |||
HIV = human immunodeficiency virus; IgA = immunoglobulin A; NSAID = nonsteroidal anti-inflammatory drug; RBC = red blood cell; WBC = white blood cell.
*—Basic laboratory work-up for suspected renal disease includes a complete blood count and measurement of serum electrolyte, blood urea nitrogen, and serum creatinine levels. Additional laboratory tests listed in this table can help evaluate for specific conditions.
†—Distinguishing among the different types of glomerulopathy is difficult clinically; therefore, they are generally diagnosed histologically.
‡—Nephrotic features: heavy proteinuria, edema, hypoalbuminemia, and hyperlipidemia; nephritic features: hematuria, hypertension, oliguria, and active urinary sediments.
§—Also a rare cause in glomerular proteinuria.
LABORATORY TESTS
A complete blood count and serum electrolyte, blood urea nitrogen, and serum creatinine measurements should be considered when appropriate if renal disease is suspected. An elevation in blood urea nitrogen or serum creatinine suggests impaired renal function. Additional blood work should be ordered when indicated by history, physical examination, or initial laboratory results (Table 2).2,19,21,22 Hematuria may be secondary to simple urinary tract infections, but may also be caused by more serious renal diseases.
IMAGING STUDIES
Ultrasonography of the urinary tract is an appropriate, noninvasive screening test for anatomic abnormalities and should be considered in patients with chronic kidney disease.2 A dimercaptosuccinic acid scan is the preferred study to detect renal scars.
RENAL BIOPSY
Renal biopsy is not routinely indicated in the proteinuria work-up.33 A biopsy should be considered when proteinuria is accompanied by active urinary sediments, persistent and gross hematuria, hypertension, hypocomplementemia, renal insufficiency with depressed glomerular filtration rate (less than 60 mL per minute per 1.73 m2 for more than three months), or signs and symptoms suggestive of vasculitic disease.34 A renal biopsy should also be considered in selected patients with nephrotic syndrome associated with a later age of onset or unresponsiveness to corticosteroid treatment.
Management
The family can be reassured if the proteinuria is transient or orthostatic, and the child is asymptomatic, has no associated hematuria, and has normal blood pressure and glomerular filtration rate. Regular follow-up is important, however, as long as significant proteinuria persists. Although there are no formal guidelines for monitoring, a child with persistent proteinuria should initially receive a physical examination, blood pressure measurement, urinalysis, and blood tests for creatinine and urea nitrogen levels every six to 12 months.29 There is no specific limitation on diet or physical activity. Once the child is stable, follow-up can be annual.
The treatment of persistent proteinuria should be directed at the underlying cause.2,29 Patients with idiopathic nephrotic syndrome should receive a trial of prednisone (2 mg per kg per day, or 60 mg per m2 per day to a maximum of 80 mg per day) in up to three divided doses for four to six weeks, followed by treatment on alternate days for another four to six weeks.35 If steroid therapy fails or adverse effects are intolerable, second-line therapy (e.g., cyclophosphamide, chlorambucil [Leukeran], cyclosporine [Sandimmune]) may be required.36 In patients with renal dysfunction, an adjunctive angiotensin-converting enzyme inhibitor and/or angiotensin-II receptor blocker can be used to decrease proteinuria and slow progression of renal disease.37–40 Referral to a pediatric nephrologist may be needed for a definitive diagnosis or consideration of renal biopsy.2
editor's note: This article is based on content that Dr. Leung is preparing for a chapter in a book titled Common Problems in Ambulatory Pediatrics. The book will be published by Nova Science Publishers, Inc., Hauppauge, NY.
