Hemolytic anemia is defined by the premature destruction of red blood cells, and can be chronic or life-threatening. It should be part of the differential diagnosis for any normocytic or macrocytic anemia. Hemolysis may occur intravascularly, extravascularly in the reticuloendothelial system, or both. Mechanisms include poor deformability leading to trapping and phagocytosis, antibody-mediated destruction through phagocytosis or direct complement activation, fragmentation due to microthrombi or direct mechanical trauma, oxidation, or direct cellular destruction. Patients with hemolysis may present with acute anemia, jaundice, hematuria, dyspnea, fatigue, tachycardia, and possibly hypotension. Laboratory test results that confirm hemolysis include reticulocytosis, as well as increased lactate dehydrogenase, increased unconjugated bilirubin, and decreased haptoglobin levels. The direct antiglobulin test further differentiates immune causes from nonimmune causes. A peripheral blood smear should be performed when hemolysis is present to identify abnormal red blood cell morphologies. Hemolytic diseases are classified into hemoglobinopathies, membranopathies, enzymopathies, immune-mediated anemias, and extrinsic nonimmune causes. Extrinsic nonimmune causes include the thrombotic microangiopathies, direct trauma, infections, systemic diseases, and oxidative insults. Medications can cause hemolytic anemia through several mechanisms. A rapid onset of anemia or significant hyperbilirubinemia in the neonatal period should prompt consideration of a hemolytic anemia.
Hemolytic anemia is defined as the destruction of red blood cells (RBCs) before their normal 120-day life span. It includes many separate and diverse entities whose common clinical features can aid in the identification of hemolysis. Hemolytic anemia exists on a spectrum from chronic to life-threatening, and warrants consideration in all patients with unexplained normocytic or macrocytic anemia.
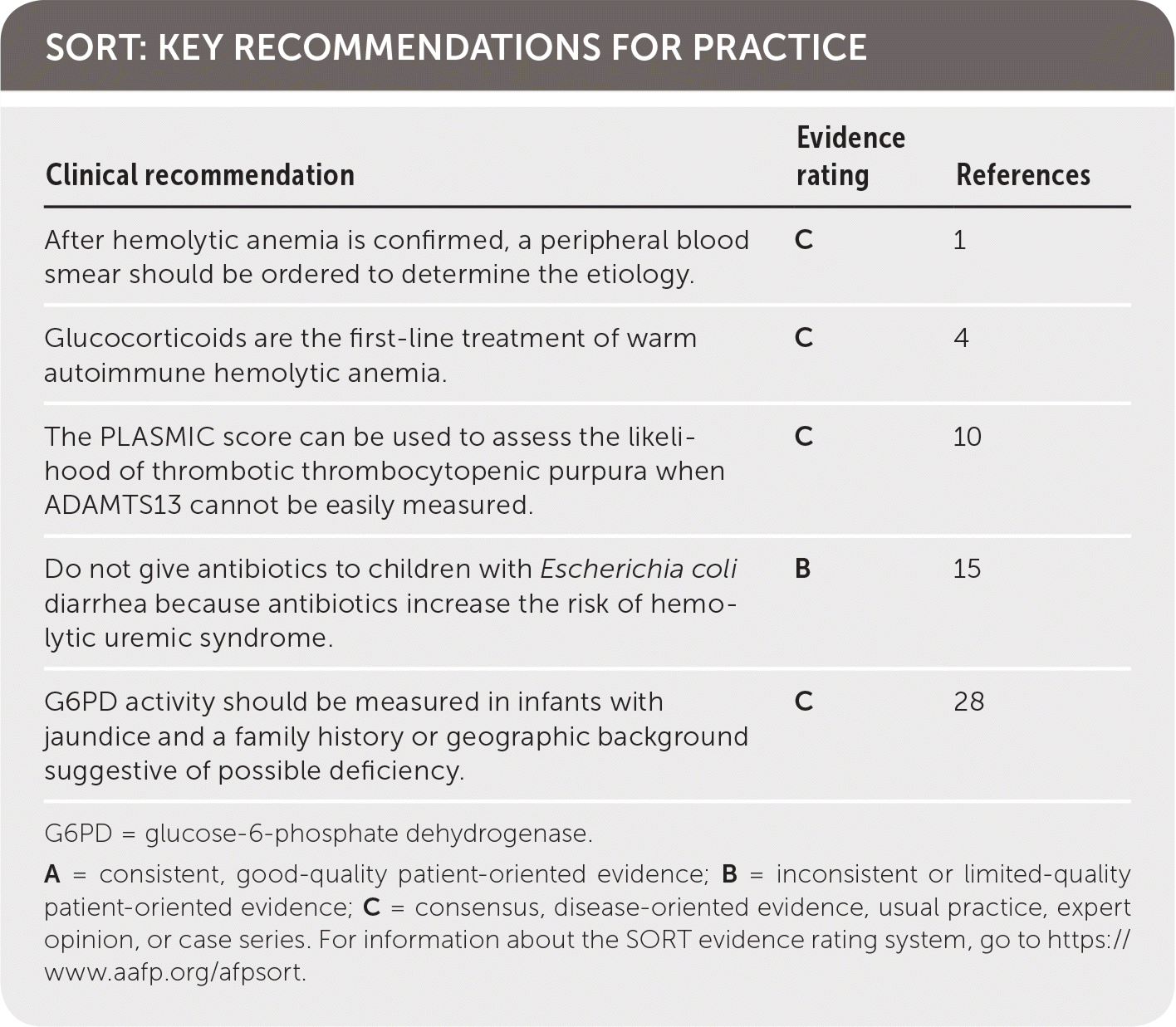
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| After hemolytic anemia is confirmed, a peripheral blood smear should be ordered to determine the etiology. | C | 1 |
| Glucocorticoids are the first-line treatment of warm autoimmune hemolytic anemia. | C | 4 |
| The PLASMIC score can be used to assess the likelihood of thrombotic thrombocytopenic purpura when ADAMTS13 cannot be easily measured. | C | 10 |
| Do not give antibiotics to children with Escherichia coli diarrhea because antibiotics increase the risk of hemolytic uremic syndrome. | B | 15 |
| G6PD activity should be measured in infants with jaundice and a family history or geographic background suggestive of possible deficiency. | C | 28 |
G6PD = glucose-6-phosphate dehydrogenase.
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.
Pathophysiology
Premature destruction of RBCs can occur intravascularly or extravascularly in the reticuloendothelial system, although the latter is more common. The primary extravascular mechanism is sequestration and phagocytosis due to poor RBC deformability (i.e., the inability to change shape enough to pass through the spleen). Antibody-mediated hemolysis results in phagocytosis or complement-mediated destruction, and can occur intravascularly or extravascularly. The intravascular mechanisms include direct cellular destruction, fragmentation, and oxidation. Direct cellular destruction is caused by toxins, trauma, or lysis. Fragmentation hemolysis occurs when extrinsic factors produce shearing and rupture of RBCs. Oxidative hemolysis occurs when the protective mechanisms of the cells are overwhelmed.1
The etiologies of hemolysis are numerous (Table 1). The hemoglobinopathies lead to splenic destruction and, in the case of sickle cell disease, likely multiple mechanisms of destruction. Inherited protein deficits lead to increased destruction in membranopathies. Enzymopathies result in hemolysis due to overwhelming oxidative stress or decreased energy production. In immune-mediated hemolytic anemia, antibodies bind with the RBCs, resulting in phagocytosis or complement-mediated destruction. The extrinsic nonimmune causes include microangiopathic hemolytic anemia (MAHA), infections, direct trauma, and drug-induced hemolysis, among others.
TABLE 1. Differential Diagnosis of Hemolysis
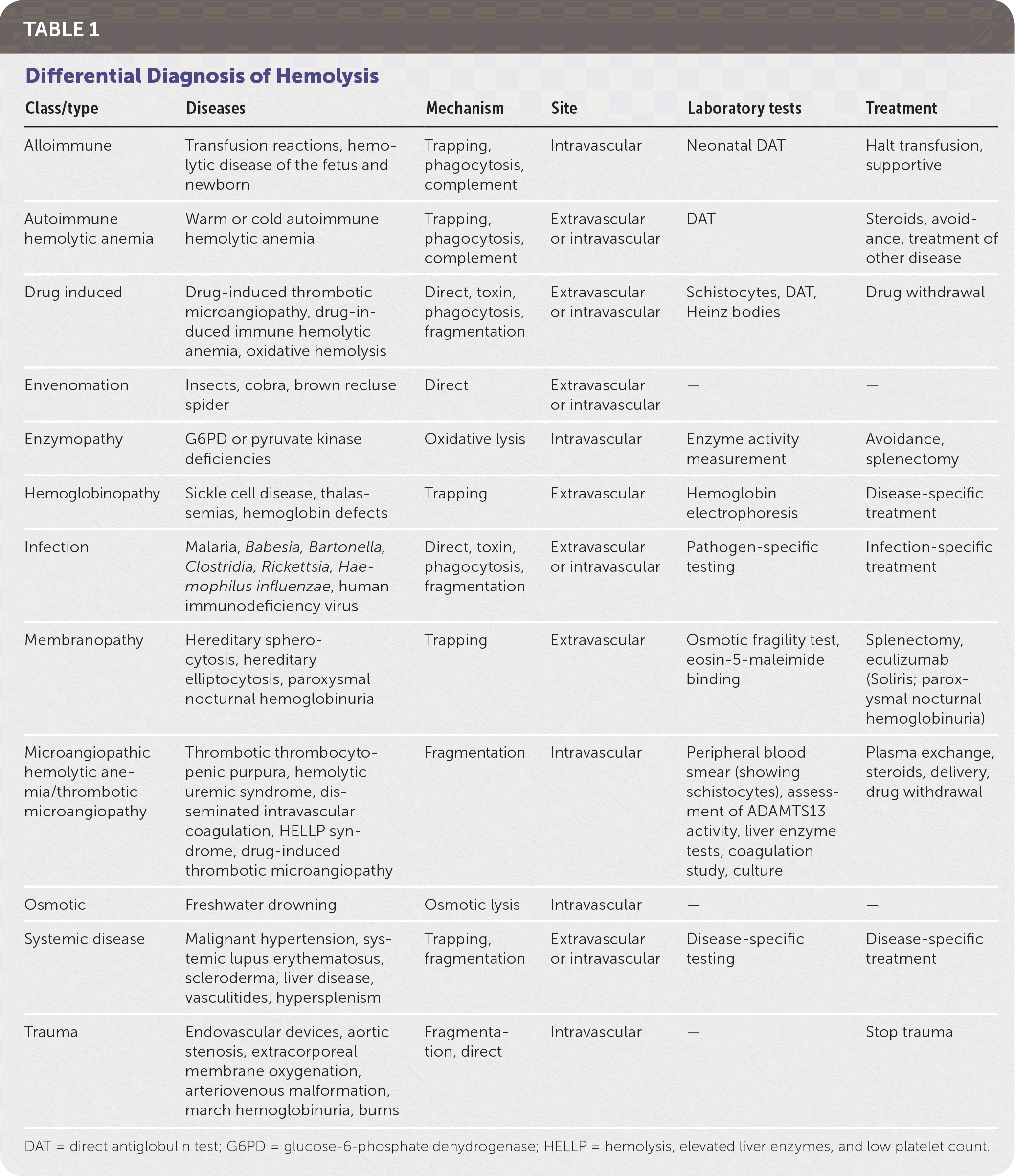
| Class/type | Diseases | Mechanism | Site | Laboratory tests | Treatment |
|---|---|---|---|---|---|
| Alloimmune | Transfusion reactions, hemolytic disease of the fetus and newborn | Trapping, phagocytosis, complement | Intravascular | Neonatal DAT | Halt transfusion, supportive |
| Autoimmune hemolytic anemia | Warm or cold autoimmune hemolytic anemia | Trapping, phagocytosis, complement | Extravascular or intravascular | DAT | Steroids, avoidance, treatment of other disease |
| Drug induced | Drug-induced thrombotic microangiopathy, drug-induced immune hemolytic anemia, oxidative hemolysis | Direct, toxin, phagocytosis, fragmentation | Extravascular or intravascular | Schistocytes, DAT, Heinz bodies | Drug withdrawal |
| Envenomation | Insects, cobra, brown recluse spider | Direct | Extravascular or intravascular | — | — |
| Enzymopathy | G6PD or pyruvate kinase deficiencies | Oxidative lysis | Intravascular | Enzyme activity measurement | Avoidance, splenectomy |
| Hemoglobinopathy | Sickle cell disease, thalassemias, hemoglobin defects | Trapping | Extravascular | Hemoglobin electrophoresis | Disease-specific treatment |
| Infection | Malaria, Babesia, Bartonella, Clostridia, Rickettsia, Haemophilus influenzae, human immunodeficiency virus | Direct, toxin, phagocytosis, fragmentation | Extravascular or intravascular | Pathogen-specific testing | Infection-specific treatment |
| Membranopathy | Hereditary spherocytosis, hereditary elliptocytosis, paroxysmal nocturnal hemoglobinuria | Trapping | Extravascular | Osmotic fragility test, eosin-5-maleimide binding | Splenectomy, eculizumab (Soliris; paroxysmal nocturnal hemoglobinuria) |
| Microangiopathic hemolytic anemia/thrombotic microangiopathy | Thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, disseminated intravascular coagulation, HELLP syndrome, drug-induced thrombotic microangiopathy | Fragmentation | Intravascular | Peripheral blood smear (showing schistocytes), assessment of ADAMTS13 activity, liver enzyme tests, coagulation study, culture | Plasma exchange, steroids, delivery, drug withdrawal |
| Osmotic | Freshwater drowning | Osmotic lysis | Intravascular | — | — |
| Systemic disease | Malignant hypertension, systemic lupus erythematosus, scleroderma, liver disease, vasculitides, hypersplenism | Trapping, fragmentation | Extravascular or intravascular | Disease-specific testing | Disease-specific treatment |
| Trauma | Endovascular devices, aortic stenosis, extracorporeal membrane oxygenation, arteriovenous malformation, march hemoglobinuria, burns | Fragmentation, direct | Intravascular | — | Stop trauma |
DAT = direct antiglobulin test; G6PD = glucose-6-phosphate dehydrogenase; HELLP = hemolysis, elevated liver enzymes, and low platelet count.
Clinical Presentation
Hemolysis should be considered when a patient experiences acute jaundice or hematuria in the presence of anemia. Symptoms of chronic hemolysis include lymphadenopathy, hepatosplenomegaly, cholestasis, and choledocholithiasis. Other nonspecific symptoms include fatigue, dyspnea, hypotension, and tachycardia.
Evaluation
When hemolysis is suspected, the history should include known medical diagnoses, medications, personal or family history of hemolytic anemia, and a complete review of systems. The physical examination should focus on identifying associated conditions, such as infections or malignancies (Table 2).
TABLE 2. Diagnostic Clues for Hemolytic Anemia
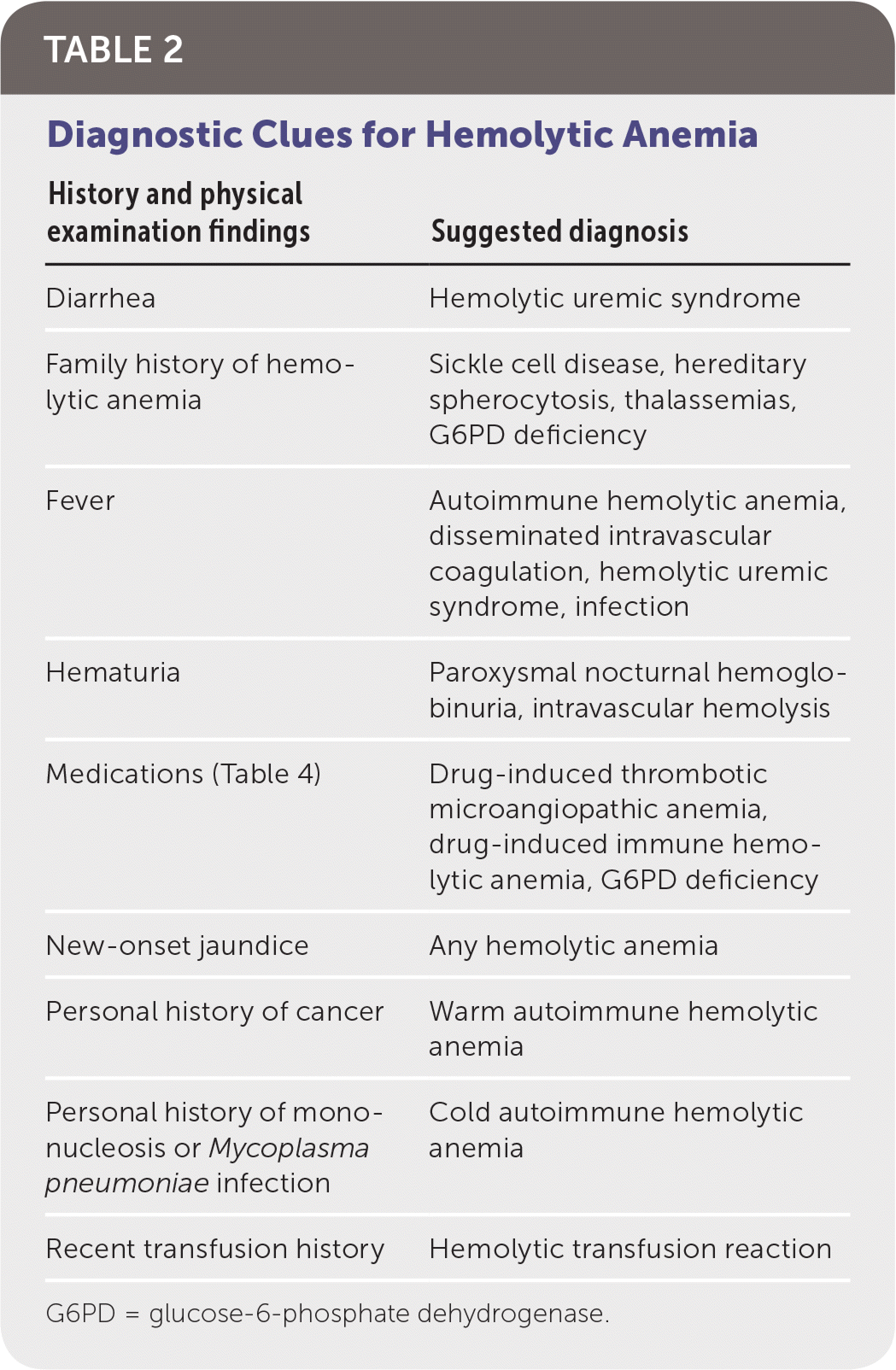
| History and physical examination findings | Suggested diagnosis |
|---|---|
| Diarrhea | Hemolytic uremic syndrome |
| Family history of hemolytic anemia | Sickle cell disease, hereditary spherocytosis, thalassemias, G6PD deficiency |
| Fever | Autoimmune hemolytic anemia, disseminated intravascular coagulation, hemolytic uremic syndrome, infection |
| Hematuria | Paroxysmal nocturnal hemoglobinuria, intravascular hemolysis |
| Medications (Table 4) | Drug-induced thrombotic microangiopathic anemia, drug-induced immune hemolytic anemia, G6PD deficiency |
| New-onset jaundice | Any hemolytic anemia |
| Personal history of cancer | Warm autoimmune hemolytic anemia |
| Personal history of mononucleosis or Mycoplasma pneumoniae infection | Cold autoimmune hemolytic anemia |
| Recent transfusion history | Hemolytic transfusion reaction |
G6PD = glucose-6-phosphate dehydrogenase.
The initial workup of hemolytic anemia begins with a complete blood count illustrating normocytic (mean corpuscular volume of 80 to 100 μm3 [80 to 100 fL]) or macrocytic (mean corpuscular volume greater than 100 μm3) anemia (Figure 1). When anemia is identified, testing should include measurement of lactate dehydrogenase, haptoglobin, reticulocyte, and unconjugated bilirubin levels, as well as urinalysis (Table 3). Lactate dehydrogenase is intracellular, and levels increase when RBCs rupture. Haptoglobin binds to free hemoglobin, and levels decrease in hemolysis. Unconjugated bilirubin levels rise as its production exceeds elimination capability. Hemolysis usually induces a reticulocytosis causing macrocytosis, unless significant iron deficiency or marrow suppression is present. Urinalysis may be positive for hemoglobinuria in hemolytic anemia despite no visible RBCs on microscopy. The constellation of reticulocytosis, increased lactate dehydrogenase levels, increased unconjugated bilirubin levels, and decreased haptoglobin levels confirms hemolysis. The absence of these findings should prompt a search for other causes. Supportive care should be initiated as required after hemolysis is confirmed.
FIGURE 1.
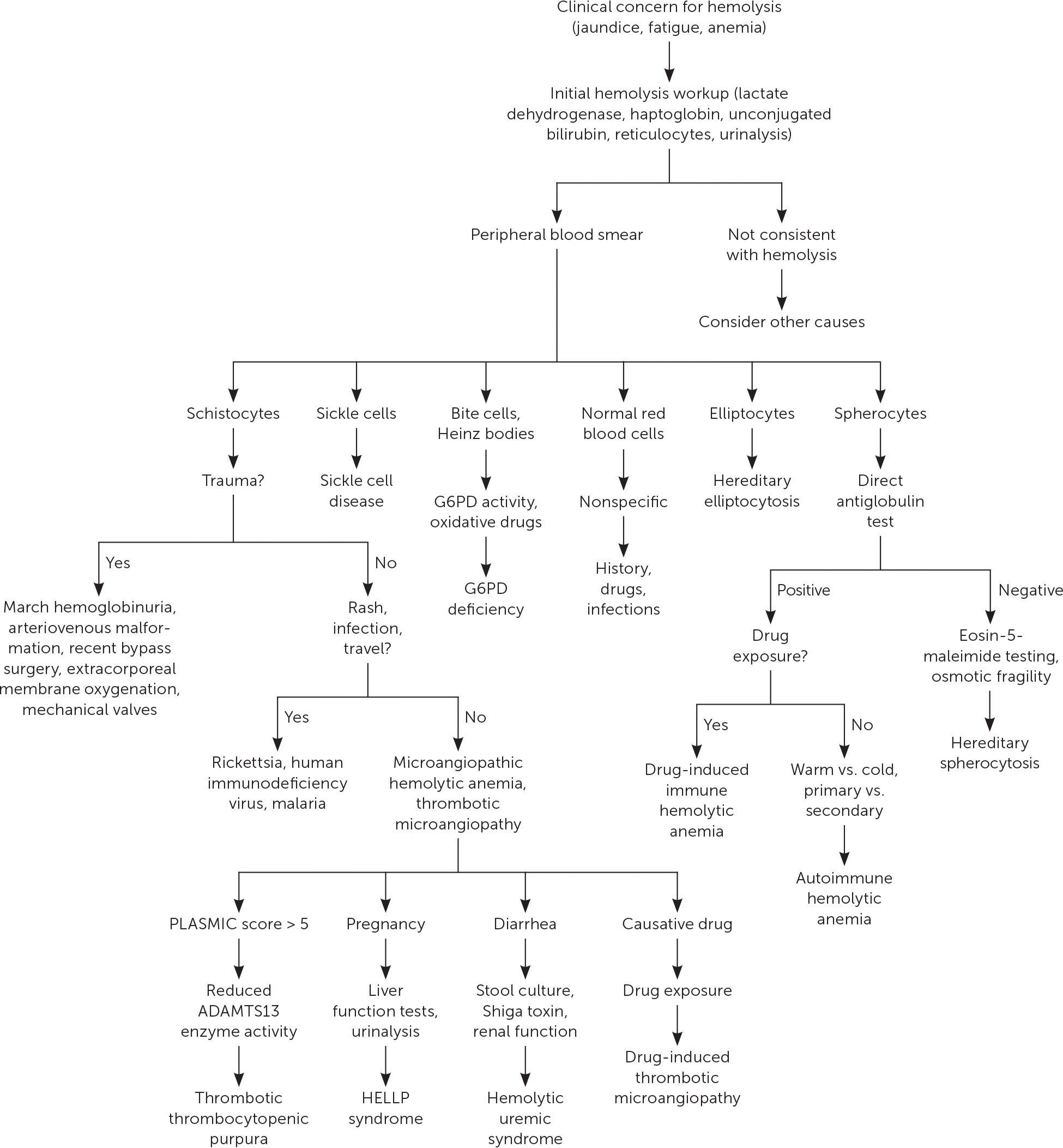
Evaluation of suspected hemolysis.
G6PD = glucose-6-phosphate dehydrogenase; HELLP = hemolysis, elevated liver enzymes, and low platelet count.
TABLE 3. Initial Laboratory Tests for Hemolysis

| Test | Finding in hemolysis | Cause |
|---|---|---|
| Haptoglobin | Decreased | Binds free hemoglobin |
| Lactate dehydrogenase | Elevated | Released from lysis of red blood cells |
| Peripheral blood smear | Abnormal red blood cells | Based on cause of anemia |
| Reticulocyte count | Increased | Marrow response to anemia |
| Unconjugated bilirubin | Increased | Increased hemoglobin breakdown |
| Urinalysis | Urobilinogen, positive for blood | Free hemoglobin and its metabolites |
Identifying the specific etiology of hemolytic anemia begins with a peripheral blood smear for abnormal RBCs, such as spherocytes, schistocytes, or bite or blister cells1 (Figure 2 and Figure 3). Spherocytes are caused by membrane deficits or repeated small membrane removals by macrophages. Spherocytosis is not diagnostic for hemolytic anemia because both hereditary spherocytosis and immune etiologies (e.g., autoimmune hemolytic anemia [AIHA], drug-induced immune hemolytic anemia) may cause spherocytes. Schistocytes are fragmented cells that result from intravascular destruction, which occurs in MAHA syndromes. Bite and blister cells result from partial phagocytosis, and occur in oxidative causes, such as glucose-6-phosphate dehydrogenase (G6PD) deficiency.2 The direct antiglobulin test (DAT) further differentiates immune causes of hemolytic anemia from nonimmune causes.
FIGURE 2.
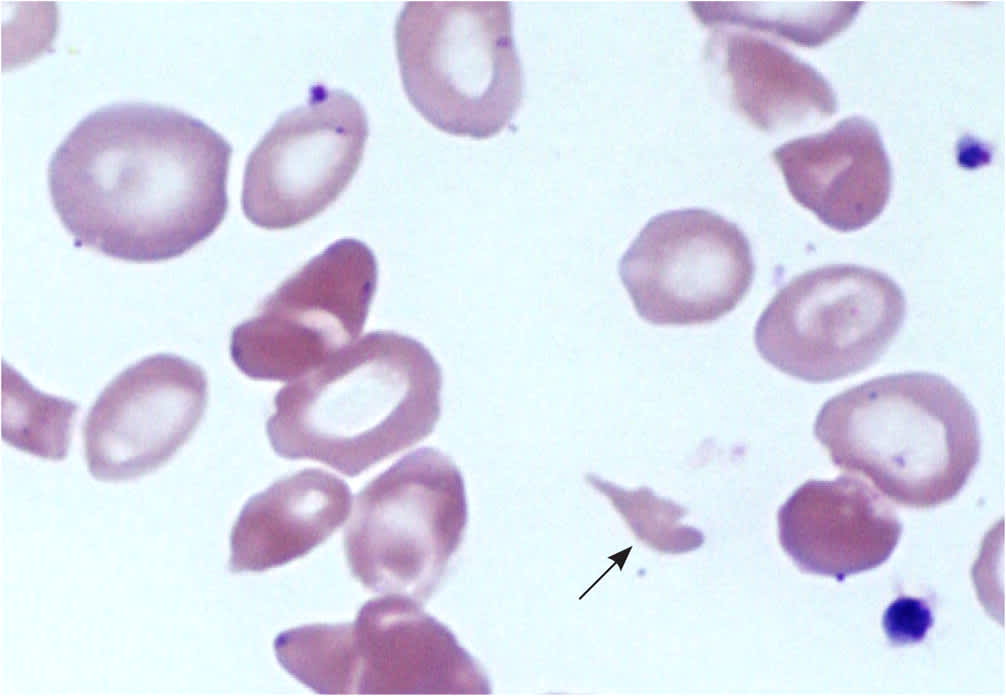
Wright-Giemsa peripheral blood smear (1,000 ×). A schistocyte (arrow) is present in the center of the image.
FIGURE 3.
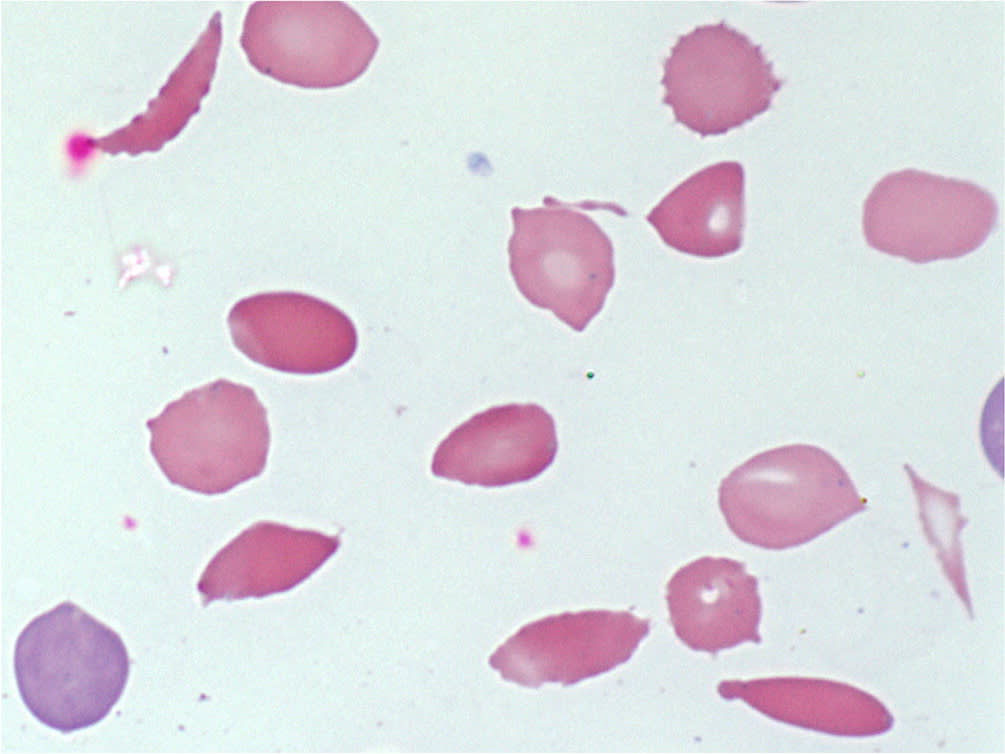
Wright-Giemsa peripheral blood smear (1,000 ×). Sickle cells, schistocytes, and acanthocytes. Lower left shows a polychromatic erythrocyte that may represent a reticulocyte (a supravital stain is needed to confirm).
Immune Hemolytic Anemia
AUTOIMMUNE HEMOLYTIC ANEMIA
AIHA is caused by autoantibody-mediated destruction. The hallmark of AIHA is a positive DAT result (Figure 4).3 AIHA is organized into two primary subgroups based on binding temperatures, referred to as cold and warm agglutinins. Many causes of AIHA are idiopathic; however, viral and bacterial infections, autoimmune conditions, connective tissue disorder, lymphoproliferative malignancies, blood transfusions, and transplantations have been associated with AIHA.
FIGURE 4.
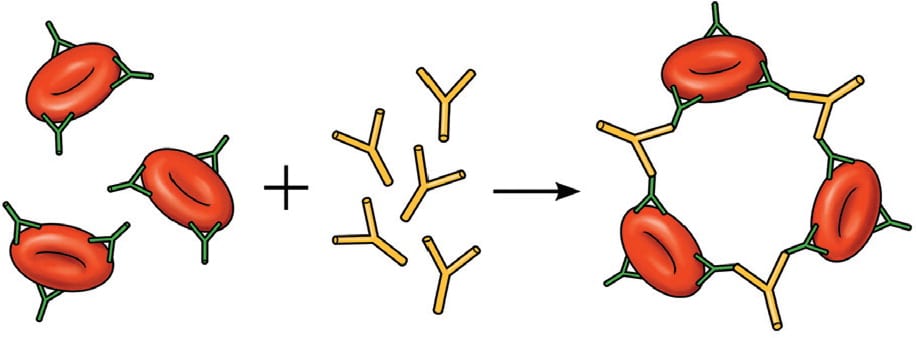
Direct antiglobulin test, demonstrating the presence of autoantibodies (shown here) or complement on the surface of the red blood cell.
Illustration by Dave Klemm
Reprinted with permission from Dhaliwal G, Cornett PA, Tierney LM Jr. Hemolytic anemia. Am Fam Physician. 2004;69(11):2602.
Warm AIHA is more common than cold AIHA and involves immunoglobulin G (IgG) antibodies, usually to the Rh complex, that react with the RBC membrane at normal body temperatures. The IgG-coated RBCs are then removed by reticuloendothelial macrophages and sequestered in the spleen, sometimes leading to splenomegaly. Treatment of warm AIHA typically includes the use of glucocorticoids, management of the underlying condition, blood transfusion (if necessary), and supportive care.4
Cold AIHA involves IgM antibodies (cold agglutinin titers) that react with polysaccharide antigens on the RBC surface at low temperatures and then cause lysis on rewarming by complement fixation and intravascular hemolysis. Development of these antibodies is associated with infectious or malignant processes. Mycoplasmal pneumonia and mononucleosis are the two most common processes. Treatment of patients who have cold AIHA typically involves supportive measures, avoidance of triggers, and underlying disease management.
TRANSFUSION REACTIONS
Acute transfusion reactions result from alloantibodies that react with incompatible RBCs. Hemolysis can range from acute to delayed, and can be life-threatening. Transfusion reactions have been discussed previously in American Family Physician.5
DRUG-INDUCED IMMUNE HEMOLYTIC ANEMIA
Drug-induced immune hemolytic anemia is a rare occurrence that results from drug-induced antibodies. A DAT result is positive in patients with this condition. Historically, methyldopa and penicillin are classic causes, but cefotetan (Cefotan), ceftriaxone, piperacillin (in combination piperacillin/tazobactam [Zosyn]), and nonsteroidal anti-inflammatory drugs currently predominate6,7 (Table 4). The progression of the condition is typically gradual, and treatment involves removal of the offending agent.
TABLE 4. Hemolysis-Inducing Agents

| Mechanism | Drugs |
|---|---|
| Drug-induced immune hemolytic anemia | Beta lactamase inhibitors, cefotetan (Cefotan), ceftriaxone, fludarabine, intravenous immunoglobulin, methlydopa, nonsteroidal anti-inflammatory drugs, penicillin, piperacillin |
| Drug-induced thrombotic microangiopathic anemia | 3,4-methylenedioxymethamphetamine (Ecstasy), bupropion (Wellbutrin), chemotherapy, clopidogrel (Plavix), cocaine, cyclosporine (Sandimmune), ibuprofen, interferon, mefloquine, metronidazole (Flagyl), nitrofurantoin, quetiapine (Seroquel), quinine, simvastatin (Zocor), tacrolimus (Prograf), trimethoprim/sulfamethoxazole, |
| Oxidation | Dapsone, nitrofurantoin, phenazopyridine, primaquine, recreational nitrates, ribavirin, rifampin |
Microangiopathic Hemolytic Anemia
MAHA is a descriptive term for hemolytic anemia that occurs when RBCs fragment, and results in schistocytes visible on the peripheral blood smear. This can be caused by trauma from an endovascular device or microthrombi. Thrombotic microangiopathies (TMAs) are a diverse group of clinical entities that share MAHA as a central feature (Table 1).
THROMBOTIC THROMBOCYTOPENIC PURPURA
Thrombotic thrombocytopenic purpura (TTP) is characterized by significantly reduced ADAMTS13 enzyme activity. The ADAMTS13 enzyme cleaves von Willebrand factor aggregations and, when it is not present or functional, large von Willebrand factor multimers form. These multimers trap platelets, causing microthrombi and RBC destruction by shearing, creating schistocytes. Approximately 95% of TTP cases are associated with acquired autoantibodies, often without an inciting event or disorder.8,9
TTP is life-threatening and requires timely diagnosis and treatment. Thrombocytopenia, fever, renal injury, MAHA, and neurologic dysfunction are hallmarks of TTP. Additional laboratory findings include a negative DAT result and normal coagulation testing. Assessment of ADAMTS13 enzyme activity is diagnostic for TTP, but results usually are delayed, making a presumptive diagnosis imperative. The PLASMIC score can be used to predict severely reduced ADAMTS13 enzyme activity and initiate early treatment (Table 5).10 Once a presumptive diagnosis is made, treatment with plasma exchange and glucocorticoids should begin immediately. Plasma exchange removes affected platelets and autoantibodies while replenishing ADAMTS13 enzyme levels. Although plasma exchange is superior, fresh frozen plasma infusion is beneficial and should be started if transfer to a plasma exchange–capable center is delayed.11
TABLE 5. PLASMIC Score for Predicting ADAMTS13 Enzyme Activity
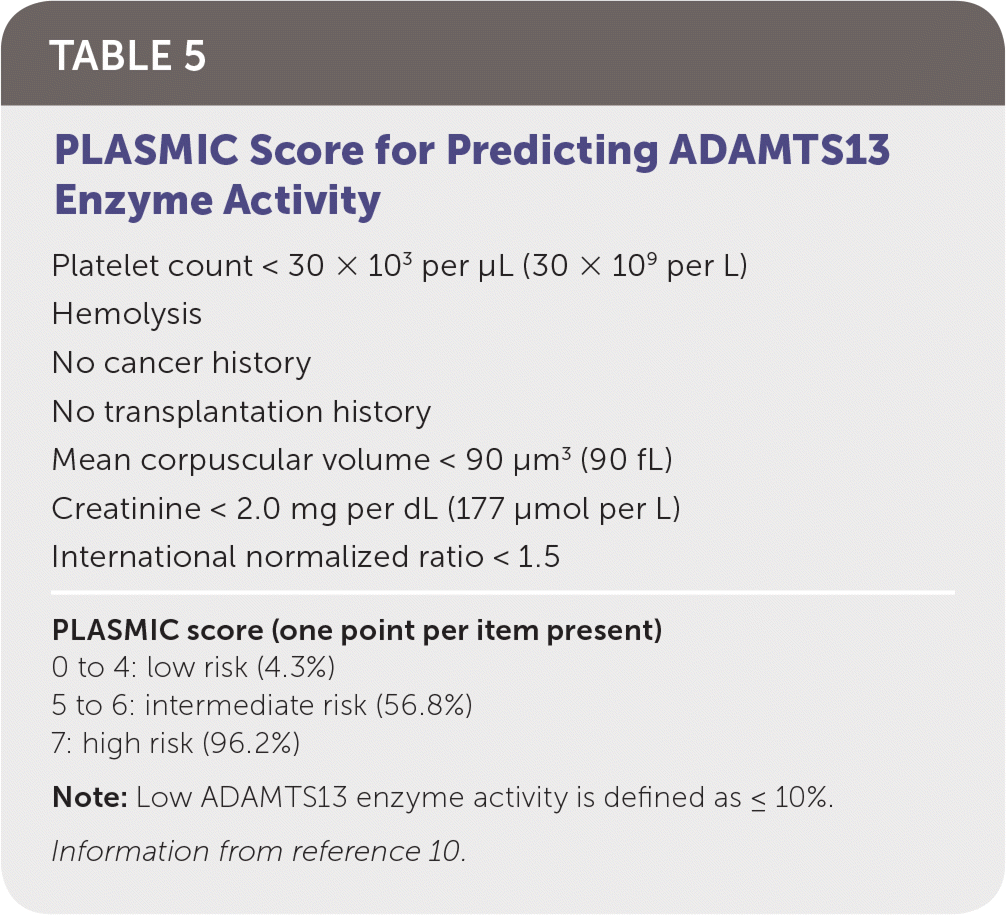
| Platelet count < 30 × 103 per μL (30 × 109 per L) |
| Hemolysis |
| No cancer history |
| No transplantation history |
| Mean corpuscular volume < 90 μm3 (90 fL) |
| Creatinine < 2.0 mg per dL (177 μmol per L) |
| International normalized ratio < 1.5 |
| PLASMIC score (one point per item present) |
| 0 to 4: low risk (4.3%) |
| 5 to 6: intermediate risk (56.8%) |
| 7: high risk (96.2%) |
Note: Low ADAMTS13 enzyme activity is defined as ≤ 10%.
Information from reference 10.
HEMOLYTIC UREMIC SYNDROME
Hemolytic uremic syndrome (HUS) is characterized by MAHA and acute kidney injury, and commonly thrombocytopenia and neurologic dysfunction. HUS is a separate entity from TTP based on ADAMTS13 enzyme activity. Shiga toxin–producing Escherichia coli HUS (STEC-HUS, also known as typical HUS) accounts for 90% of HUS cases and is caused by STEC organisms such as O157:H7 and Shigella dysenteriae.12 It predominantly affects children. STEC-HUS has a classic prodrome of abdominal pain with diarrhea, often preceding MAHA, acute kidney injury, and thrombocytopenia by five to 10 days. STEC urinary tract infections can cause a diarrhea-negative STEC-HUS.13 Streptococcus pneumoniae, human immunodeficiency virus, and influenza have also been associated with HUS in rare cases, which presents without the classic prodrome. There is an atypical HUS that also does not have the prodrome, is caused by complement dysregulation and not infection, and can be hereditary. Exacerbations can be triggered by an upper respiratory tract infection.14
Inadequately cooked ground beef is the primary source of STEC infection, but fruits, vegetables, poultry, and contaminated drinking water have also been implicated. In STEC-HUS, the Shiga toxin is absorbed and attaches to specific receptors, most expressed in the glomerulus and brain in children, causing endothelial cell damage, which initiates a cascade resulting in large von Willebrand factor multimers that induce MAHA. Treatment of STEC-HUS is supportive care and continued evaluation of renal function. Antibiotics are not recommended for gastrointestinal STEC because they may increase the risk of HUS.15
OTHER MICROANGIOPATHIC HEMOLYTIC ANEMIA SYNDROMES
Other clinical entities that cause MAHA include HELLP syndrome (hemolysis, elevated liver enzymes, and low platelet count) and disseminated intravascular coagulation. HELLP syndrome is pregnancy related and shares many characteristics with TTP and HUS, both of which can also occur during pregnancy. TTP and HUS do not usually induce liver-associated enzyme elevations as in HELLP syndrome.16 A low lactate dehydrogenase–to–aspartate transaminase ratio can aid in distinguishing HELLP syndrome, because the rate of hemolysis is higher in the other TMAs and hepatic involvement is higher in HELLP syndrome.17 Disseminated intravascular coagulation also can result in MAHA due to fibrin-rich microthrombi. Disseminated intravascular coagulation causes prolonged coagulation studies, positive d-dimer test results, and decreased fibrinogen levels.18
DRUG-INDUCED THROMBOTIC MICROANGIOPATHY
Drug-induced TMA occurs when a compound causes the formation of platelet microthrombi, resulting in MAHA through induced antibodies or direct toxicity. These antibodies interact strongly only in the presence of the drug. The clinical features are similar to those of other MAHA syndromes. In a 2015 review, the overall incidence of drug-induced TMA was 5% of all MAHA cases.19 Another review found 78 medications suspected of causing drug-induced TMA. Quinine, cyclosporine (Sandimmune), and tacrolimus (Prograf) constituted more than one-half of all drug-induced TMA cases20 (Table 4). The management of drug-induced TMA includes discontinuing the offending agent and providing supportive care; plasma exchange is not beneficial, except in the case of ticlopidine.21
Oxidative Hemolytic Anemia
Oxidative hemolysis occurs when normal processes are unable to reduce ferric (3+) iron, also known as methemoglobin, to ferrous (2+) iron, which carries oxygen. This results in methemoglobinemia (i.e., the denaturing of ferric hemoglobin into multimers, called Heinz bodies), leading to premature RBC destruction by phagocytosis. G6PD is integral to these protective systems, and when it is deficient, oxidative insults may cause hemolysis. G6PD deficiency is an X-linked disorder and is common in individuals of Mediterranean and African descent. Classically, fava beans, sulfa drugs, and primaquine were the primary triggers of oxidative hemolysis, but the list of medications to avoid in persons with G6PD deficiency is extensive22,23 (see https://www.aafp.org/afp/2005/1001/p1277.html#afp20051001p1277-t3). The diagnosis is made by G6PD activity testing, although this may be normal during or just after a hemolytic episode.24 Amyl and butyl nitrate, topical benzocaine, phenazopyridine, dapsone, ribavirin, and paraquat ingestion can also cause oxidative hemolysis, even with normal G6PD levels (Table 4). Treatment is discontinuation of the drug and supportive care. Methylene blue is indicated for the treatment of severe methemoglobinemia from a non-G6PD cause, but it is possibly harmful and contraindicated in persons with G6PD deficiency.25
Considerations in Children
A rapid onset of anemia or significant hyperbilirubinemia (i.e., based off of physiologic norms depending on age) in the neonatal period should prompt consideration of hemolytic anemia. Hemolytic disease of the fetus and newborn is an alloimmune hemolysis caused by maternal antibodies in the neonate's plasma, is most commonly anti-Rh, and is DAT-positive. Hemolytic disease of the fetus and newborn is treated with supportive care and hyperbilirubinemia management. Severe G6PD deficiency can also cause acute hemolysis, accounting for nearly 30% of all kernicterus cases in one observational study.26 G6PD deficiency is a key part of the differential diagnosis of neonatal hyperbilirubinemia, particularly in high-incidence populations.27,28
Hereditary spherocytosis is the most common inherited membranopathy and is caused by one of several defective proteins. In severe cases, it can cause hemolysis in the neonatal period but typically presents later as chronic hemolysis. The mutations are largely autosomal dominant, making the family history important. In the setting of a DAT-negative hemolysis and spherocytes on peripheral blood smear, an increased mean corpuscular hemoglobin concentration–to–mean corpuscular volume ratio (greater than 0.35) should prompt consideration of hereditary spherocytosis and further testing with osmotic fragility and eosin-5-maleimide binding to confirm the diagnosis.29 Treatment is supportive, with splenectomy often required in moderate to severe hereditary spherocytosis.
Inherited hemoglobinopathies, such as sickle cell disease and thalassemias, can present in the neonatal period or later, depending on their severity. They should always be considered in the workup of a child with hemolysis, and have been reviewed in previous articles in American Family Physician30,31 (https://www.aafp.org/afp/2015/1215/p1069.html and https://www.aafp.org/afp/2009/0815/p339.html).
This article updates a previous article on this topic by Dhaliwal, et al.3
Data Sources: We searched PubMed with the terms MAHA, TMA, TTP, HUS, DIC, hyperbilirubinemia, and G6PD deficiency. Relevant UpToDate articles were also accessed to provide references. An Essential Evidence Plus search was provided to the authors as well. The Cochrane database was also queried. Search dates: April and May 2017, and March 2018.
Figure 2 and Figure 3 provided by Lyndon P. Bowden, MD, MPH, Department of Pathology, Womack Army Medical Center.
The views expressed in this article are those of the authors and do not necessarily reflect the official policy of the Department of the Army, the Department of Defense, or the U.S. government.
