Hereditary hemochromatosis is an autosomal recessive disorder that disrupts iron homeostasis, resulting in systemic iron overload. It is the most common inherited disorder among people of northern European ancestry. Despite the high prevalence of the gene mutation, there is a low and variable clinical penetrance. The deposition of excess iron into parenchymal cells leads to cellular dysfunction and the clinical manifestations of the disease. The liver, pancreas, joints, heart, skin, and pituitary gland are the most commonly involved organs. Hereditary hemochromatosis is usually diagnosed in the 40s or 50s. Women are often diagnosed later than men, likely because of menstrual blood loss. There is no typical presentation or pathognomonic signs and symptoms of hereditary hemochromatosis. Because of increased awareness and earlier diagnosis, the end-organ damage secondary to iron overload is not often seen in clinical practice. A common initial presentation is an asymptomatic patient with mildly elevated liver enzymes who is subsequently found to have elevated serum ferritin and transferrin saturation. Ferritin levels greater than 300 ng per mL for men and 200 ng per mL for women and transferrin saturations greater than 45% are highly suggestive of hereditary hemochromatosis. Phlebotomy is the mainstay of treatment and can help improve heart function, reduce abnormal skin pigmentation, and lessen the risk of liver complications. Liver transplantation may be considered in select patients. Individuals with hereditary hemochromatosis have an increased risk of hepatocellular carcinoma and colorectal and breast cancers. Genetic testing for the hereditary hemochromatosis genes should be offered after 18 years of age to first-degree relatives of patients with the condition.
Hereditary hemochromatosis is an autosomal recessive condition that results in systemic iron overload due to a deficiency in hepcidin, an iron regulatory protein.1,2 The body's iron stores are primarily regulated by controlling intestinal absorption. Other than menses and shedding of senescent cells, there are no physiologic mechanisms of iron excretion. Deposition of excess iron into parenchymal cells leads to tissue damage and ultimately organ failure. The liver, pancreas, joints, heart, skin, and pituitary glands are the most commonly involved organs.3,4 This article summarizes the best available patient-oriented evidence regarding hereditary hemochromatosis.
SORT: KEY RECOMMENDATIONS FOR PRACTICE
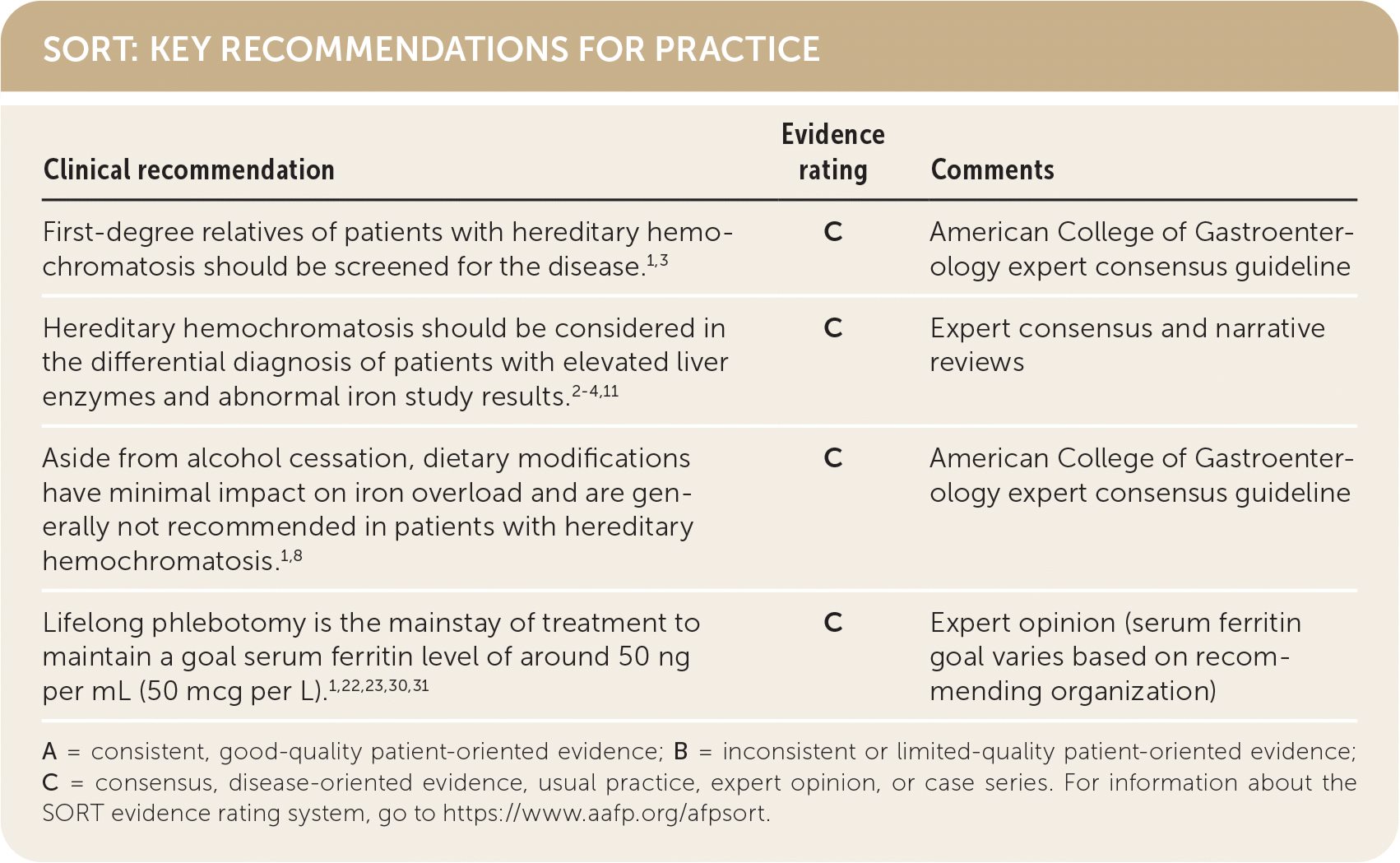
| Clinical recommendation | Evidence rating | Comments |
|---|---|---|
| First-degree relatives of patients with hereditary hemochromatosis should be screened for the disease.1,3 | C | American College of Gastroenterology expert consensus guideline |
| Hereditary hemochromatosis should be considered in the differential diagnosis of patients with elevated liver enzymes and abnormal iron study results.2–4,11 | C | Expert consensus and narrative reviews |
| Aside from alcohol cessation, dietary modifications have minimal impact on iron overload and are generally not recommended in patients with hereditary hemochromatosis.1,8 | C | American College of Gastroenterology expert consensus guideline |
| Lifelong phlebotomy is the mainstay of treatment to maintain a goal serum ferritin level of around 50 ng per mL (50 mcg per L).1,22,23,30,31 | C | Expert opinion (serum ferritin goal varies based on recommending organization) |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.
BEST PRACTICES IN GENETICS

| Recommendation | Sponsoring organization |
|---|---|
| Do not order HFE genetic testing for a patient without iron overload or a family history of HFE-associated hereditary hemochromatosis. | American College of Medical Genetics and Genomics |
Source: For more information on the Choosing Wisely Campaign, see https://www.choosingwisely.org. For supporting citations and to search Choosing Wisely recommendations relevant to primary care, see https://www.aafp.org/afp/recommendations/search.htm.
Epidemiology
- There are four types of hereditary hemochromatosis, which are categorized by the specific gene mutation involved (Table 1).1,5,6
- Homozygous C282Y and heterozygous C282Y/H63D mutations of the HFE gene (iron regulatory protein) on chromosome 6 are responsible for up to 95% of hereditary hemochromatosis cases (type 1).1,3
- Hereditary hemochromatosis types 2 to 4 comprise the small percentage of remaining cases and are caused by non- HFE mutations.1,5
- Hereditary hemochromatosis is the most common inherited disorder among people of northern European ancestry.1,3 The United States, Europe, and Australia have a similar disease prevalence of one case per 200 to 400 people, with the highest prevalence in those of Irish and Scandinavian ancestry.1,3,5
- The hereditary hemochromatosis mutation is rare in people of Asian, African, Hispanic, and Pacific Islander ancestry.7
- Despite the high prevalence of the gene mutation, there is a low and variable clinical penetrance, with up to 25% of people with C282Y homozygosity being clinically asymptomatic.4,8
- The clinical manifestations of iron overload typically develop in the 40s or 50s.1
- The disease is equally prevalent in men and women. Women typically present later in life when they are postmenopausal, likely because of menstrual blood loss delaying the development of symptomatic iron overload.1,3
TABLE 1. Hereditary Hemochromatosis Subtypes
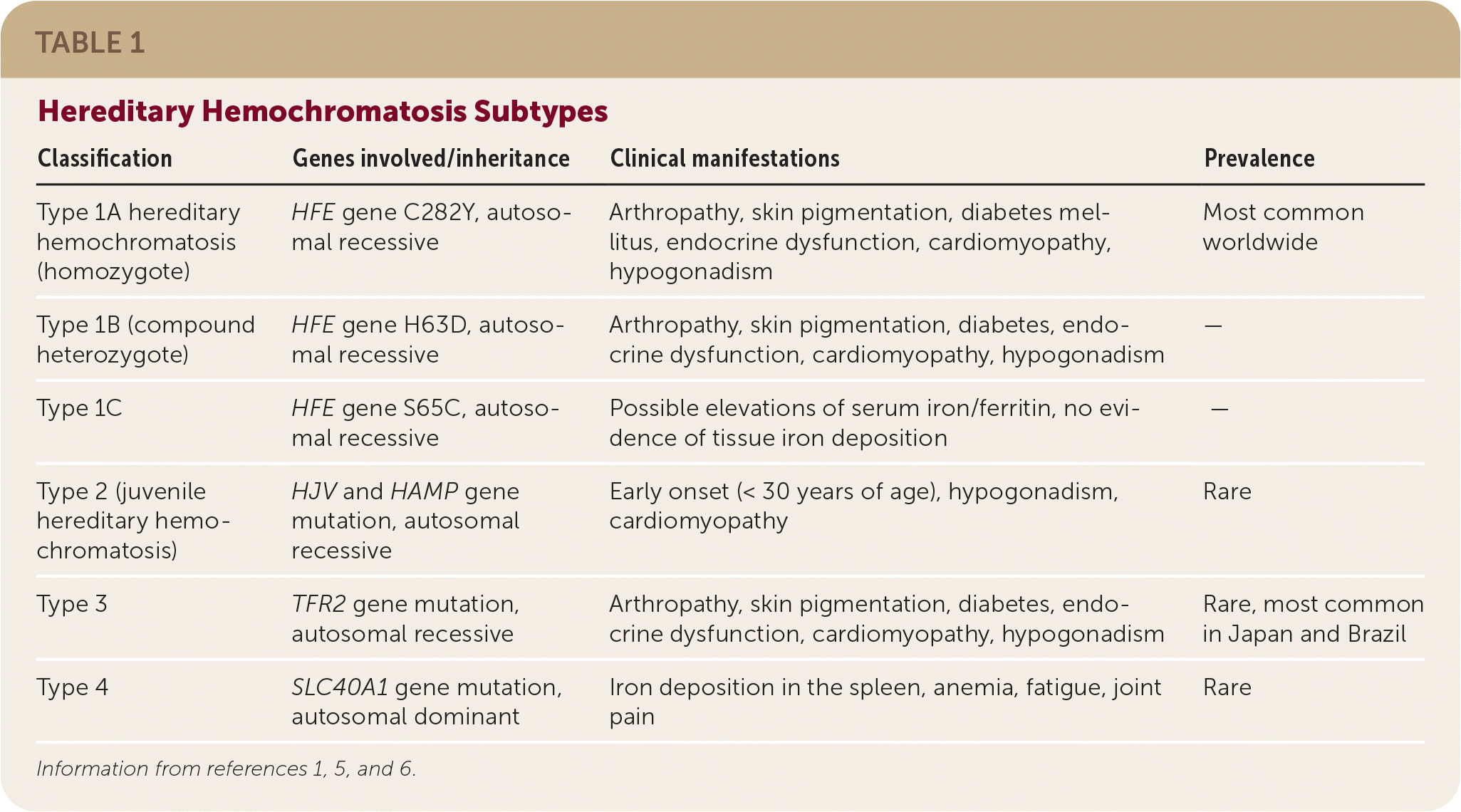
| Classification | Genes involved/inheritance | Clinical manifestations | Prevalence |
|---|---|---|---|
| Type 1A hereditary hemochromatosis (homozygote) | HFE gene C282Y, autosomal recessive | Arthropathy, skin pigmentation, diabetes mellitus, endocrine dysfunction, cardiomyopathy, hypogonadism | Most common worldwide |
| Type 1B (compound heterozygote) | HFE gene H63D, autosomal recessive | Arthropathy, skin pigmentation, diabetes, endocrine dysfunction, cardiomyopathy, hypogonadism | — |
| Type 1C | HFE gene S65C, autosomal recessive | Possible elevations of serum iron/ferritin, no evidence of tissue iron deposition | — |
| Type 2 (juvenile hereditary hemochromatosis) | HJV and HAMP gene mutation, autosomal recessive | Early onset (< 30 years of age), hypogonadism, cardiomyopathy | Rare |
| Type 3 | TFR2 gene mutation, autosomal recessive | Arthropathy, skin pigmentation, diabetes, endocrine dysfunction, cardiomyopathy, hypogonadism | Rare, most common in Japan and Brazil |
| Type 4 | SLC40A1 gene mutation, autosomal dominant | Iron deposition in the spleen, anemia, fatigue, joint pain | Rare |
Screening and Prevention
- Genetic testing for the C282Y and H63D mutations of the HFE gene is recommended for first-degree relatives of people with hereditary hemochromatosis; screening does not need to begin until 18 years of age because clinical manifestations are rare before this age.1,3
- Population-level genetic testing or laboratory screening for hereditary hemochromatosis is not recommended because of the cost and the variable prevalence and incomplete penetrance of the C282Y mutation.1,3–5,9
Diagnosis
- The presentation of hereditary hemochromatosis varies, with biochemical expression occurring first and clinical expression occurring in later stages of disease.3,8 Table 2 summarizes the stages of hereditary hemochromatosis.1,2,4,5
- The differential diagnosis includes other causes of secondary iron overload via excess absorption and organ deposition of iron (Table 3).1–5,10
- Hereditary hemochromatosis should be considered in the differential diagnosis of patients with elevated liver enzymes and abnormal iron study results.2–4,11
TABLE 2. Stages of Hereditary Hemochromatosis
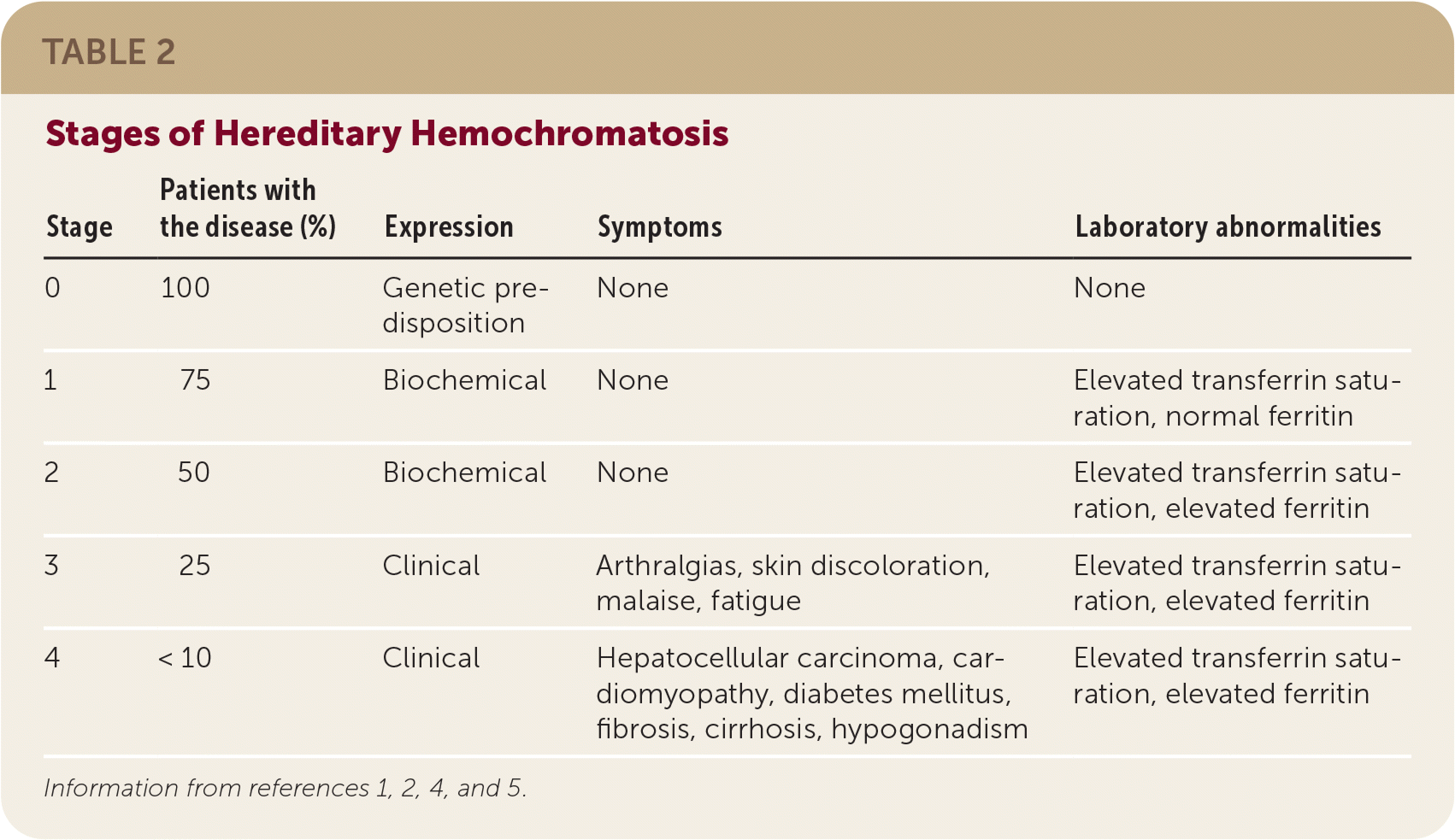
| Stage | Patients with the disease (%) | Expression | Symptoms | Laboratory abnormalities |
|---|---|---|---|---|
| 0 | 100 | Genetic predisposition | None | None |
| 1 | 75 | Biochemical | None | Elevated transferrin saturation, normal ferritin |
| 2 | 50 | Biochemical | None | Elevated transferrin saturation, elevated ferritin |
| 3 | 25 | Clinical | Arthralgias, skin discoloration, malaise, fatigue | Elevated transferrin saturation, elevated ferritin |
| 4 | < 10 | Clinical | Hepatocellular carcinoma, cardiomyopathy, diabetes mellitus, fibrosis, cirrhosis, hypogonadism | Elevated transferrin saturation, elevated ferritin |
TABLE 3. Conditions That May Present with, or Similarly to, Iron Overload

| Anemias associated with dysfunctional erythropoiesis Aplastic anemia Chronic hemolytic anemia Hereditary hemochromatosis Hereditary spherocytosis Long-term hemodialysis Pyruvate kinase deficiency Sickle cell anemia Thalassemia major Chronic liver disease Alcoholic liver disease Hepatitis B Hepatitis C Nonalcoholic fatty liver disease Porphyria cutanea tarda Inflammatory states Rheumatoid arthritis Systemic lupus erythematosus Malignancy Breast cancer Hematologic Hepatocellular carcinoma Parenteral iron overload Iron transfusion Long-term hemodialysis Red blood cell transfusion |
SIGNS AND SYMPTOMS
- Hereditary hemochromatosis has no pathognomonic signs and symptoms, with excess iron accumulating in multiple organ systems and leading to a variety of presentations (Table 4).1–5 There are no good data on the predictive value of symptoms in identifying patients at risk of hereditary hemochromatosis.1,3,8
- A common presentation is an asymptomatic patient who has abnormal hepatic transaminase levels and then elevated serum ferritin and/or transferrin saturation on subsequent testing.
- Fatigue and arthralgias are the most common early clinical symptoms, followed by decreased libido.1,3
- Hereditary hemochromatosis has classically been characterized by a triad of diabetes mellitus, liver cirrhosis, and abnormal skin pigmentation; however, this late-stage presentation is now uncommon given increased clinical awareness and advances in diagnosis and treatment.2
- End-organ damage secondary to hereditary hemochromatosis develops in 10% of those with the HFE homozygous mutation.3,4,8
TABLE 4. Signs and Symptoms of Hereditary Hemochromatosis

| Endocrine Diabetes mellitus Hyperglycemia Hypopituitarism Hypothyroidism General Apathy Fatigue Malaise Weakness Weight loss Heart Arrhythmias Cardiomegaly Heart failure Joints Arthralgias, especially in the second and third metacarpophalangeal joints Arthritis with boggy and tender joints Decreased grip strength Liver Abdominal pain, especially in the right upper quadrant Ascites Cirrhosis Elevated liver enzymes Fibrosis Hepatocellular carcinoma Hepatomegaly Reproductive organs Amenorrhea Decreased libido Impotence Testicular atrophy Skin Jaundice Skin hyperpigmentation (bronzing) Spider angiomas |
DIAGNOSTIC TESTING
- Figure 1 is a suggested approach to the evaluation of patients in the general population with suspected hereditary hemochromatosis.7,9,10 The relative accuracy of diagnostic tests for hereditary hemochromatosis is shown in Table 5.5,12–16
- Transferrin saturation of greater than 45% is often the earliest laboratory abnormality detected.7
- Ferritin levels greater than 300 ng per mL (300 mcg per L) in men and greater than 200 ng per mL (200 mcg per L) in women suggest iron overload, but other diagnoses must be ruled out before attributing them to hereditary hemochromatosis.7,17
- In the past, fasting blood samples were recommended to avoid circadian and postprandial variations in ferritin levels because nonfasting samples were thought to increase the false-positive rate. Newer data call this concept into question, and blood draws can be performed regardless of whether the patient is fasting.5,6
- Determining the hepatic iron concentration via low T2-weighted magnetic resonance imaging can help predict the long-term risk of developing cirrhosis.1
- Transient elastography is a noninvasive method for determining the extent of cirrhosis.
- Liver biopsy is reserved for staging of hepatic fibrosis or when evaluating for an alternate diagnosis.1,4
FIGURE 1.
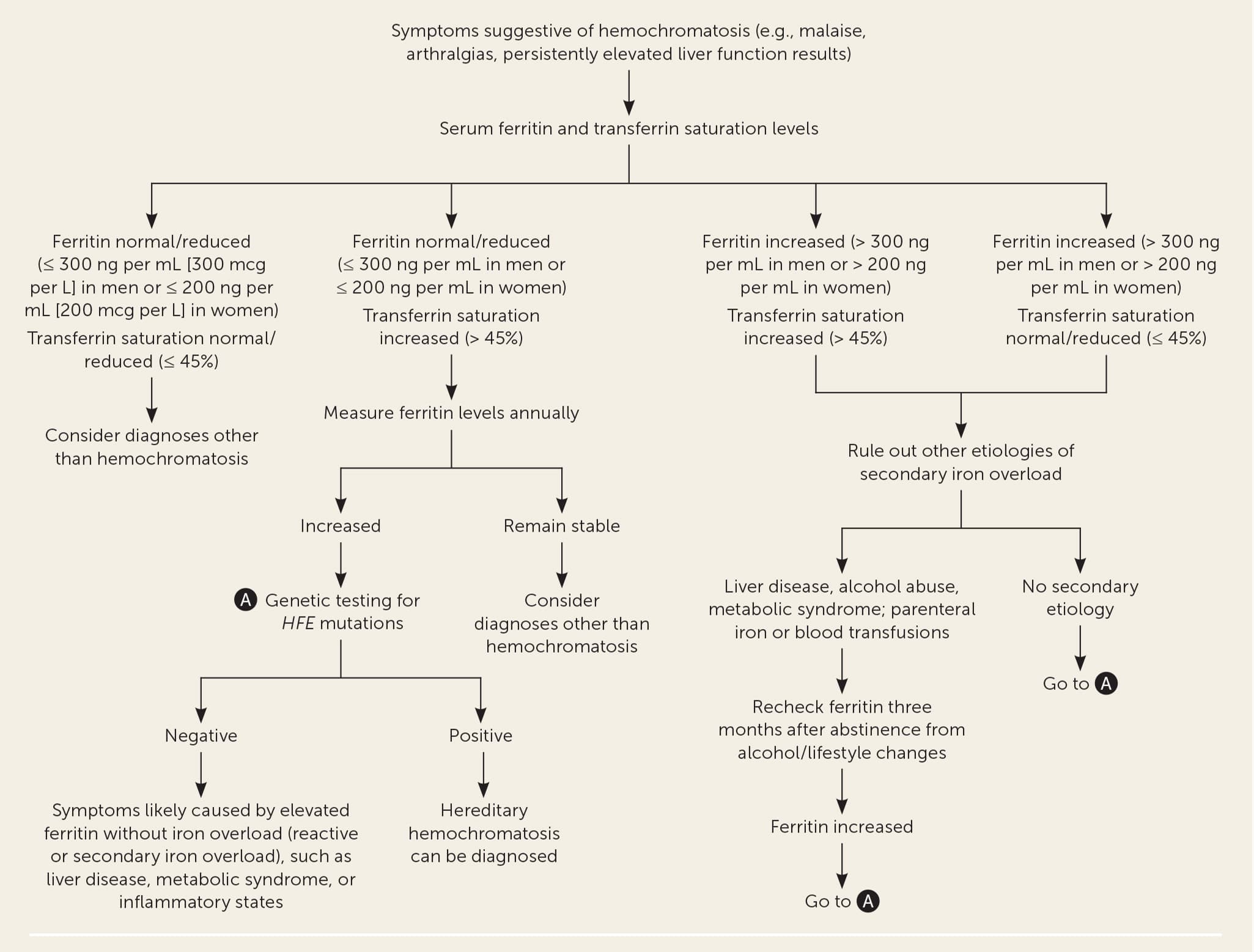
Approach to the diagnostic evaluation of suspected hereditary hemochromatosis.
TABLE 5. Accuracy of Diagnostic Tests for Hereditary Hemochromatosis

| Diagnostic test | Sensitivity | Specificity | LR+ | LR− |
|---|---|---|---|---|
| Hepatic iron index ≥ 1.912 | 93% | 100% | 186 | 0.07 |
| Transferrin saturation > 45%13 | 98% | 99% | 98 | 0.01 |
| Ferritin > 200 ng per mL (200 mcg per L) in women5,14 | 66% | 85% | 4.4 | 0.4 |
| Ferritin > 300 ng per mL (300 mcg per L) in men5,14 | 66% | 85% | 4.4 | 0.4 |
| Serum iron > 168 ng per mL15 | 68% | 83% | 4.0 | 0.4 |
| C282Y genetic testing16 | 91.3% to 92.4% | 98.8% to 100% | ~99 | ~0.08 |
LR− = negative likelihood ratio; LR+ = positive likelihood ratio.
Treatment
LIFESTYLE THERAPY
- Obesity and alcohol intake can compound the risk of liver injury in hereditary hemochromatosis. Patients with the disease should avoid alcohol and maintain a healthy body weight and level of physical activity.2,18,19
- Aside from alcohol cessation, dietary modifications have minimal impact on iron overload. Guidelines do not support specific dietary interventions or avoidance of plant-based iron sources when patients are undergoing phlebotomy, although avoiding iron-rich foods may decrease the amount of blood that needs to be phlebotomized.1,8
- Based on pathophysiologic reasoning and expert opinion, restricting vitamin C intake to 500 mg per day and reducing red meat consumption are often recommended.8,20,21
MEDICAL THERAPY
- Early detection and adequate treatment in the early stages of disease (prediabetes and prefibrosis) can result in improvement and resolution of symptoms and a normal survival rate.3
- Phlebotomy has not been studied in a randomized controlled trial, but it is considered the mainstay of treatment to remove excess iron and improve outcomes if irreversible end-organ damage has not occurred.22–24
- Phlebotomy should be offered to patients with elevated serum ferritin levels (greater than 300 ng per mL in men and greater than 200 ng per mL in women) or a transferrin saturation of greater than 45% in both men and women.
- Phlebotomy may improve heart function, reduce abnormal skin pigmentation, and lessen the risk of liver complications. It likely will not improve preexisting diabetes, arthralgias, cirrhosis, and hypogonadism.11,25–29
- The American College of Gastroenterology recommends weekly removal of 500 mL of blood, increasing or decreasing the amount based on patient tolerance.1 Monthly ferritin levels should be tracked, with weekly blood removal continuing until a serum ferritin level of 50 to 150 ng per mL (50 to 150 mcg per L) is obtained. Once this goal is achieved, the patient should undergo lifelong phlebotomy three or four times per year, with ferritin monitoring every six months, to maintain levels around 50 ng per mL.22,23,30,31
- Adverse effects of phlebotomy, such as phlebitis, malaise, hematoma, delayed bleeding, infection, neurovascular damage, and fatigue, occur in 37% to 50% of patients.32
- Erythrocytapheresis, a red blood cell apheresis treatment that removes red blood cells and returns the other blood components to the patient, is limited by its availability and cost. There is insufficient evidence to assess the benefits and harms of this treatment compared with phlebotomy.24,32
- Chelation, which uses medications to bind serum iron, is not a first-line therapy because of significant adverse effects (e.g., neurotoxicity, neutropenia, agranulocytosis, increased serum creatinine, increased liver enzymes) and limited evidence. Chelation should be considered in consultation with a specialist and only if phlebotomy is contraindicated or ineffective.30,31
- Patients with hereditary hemochromatosis should be vaccinated against hepatitis A and B, if not already immune.8
- Although not recommended in patients without other indications (e.g., gastroesophageal reflux disease), proton pump inhibitors lower the acidity of the stomach and reduce iron absorption. A systematic review and meta-analysis demonstrated a decreased need for phlebotomies in patients taking proton pump inhibitors for at least one year.33
SURGICAL THERAPY
Surveillance for Complications
- The incidence of hepatocellular carcinoma is 20 to 200 times higher in patients with hereditary hemochromatosis, especially those with higher than stage 3 fibrosis and cirrhosis.1 Hepatocellular carcinoma accounts for nearly 45% of deaths in patients with hereditary hemochromatosis.1
- Liver imaging, with or without alpha-fetoprotein measurements, should be performed every six months for life to screen for hepatocellular carcinoma and monitor for the progression of cirrhosis. This should occur even after iron removal because hepatocellular carcinoma can develop years after successful iron depletion.1,2,4
- Laboratory testing and/or liver ultrasonography should be performed in patients with hereditary hemochromatosis to screen for concomitant liver diseases, such as hepatitis A, B, and C; alcoholic liver disease; and nonalcoholic fatty liver disease.2,4
- There is a twofold increased risk of colorectal and breast cancers in people with hereditary hemochromatosis C282Y homozygosity. There does not appear to be an increased cancer risk in those with C282Y/H63D heterozygosity or a non- HFE mutation.35 Patients with hereditary hemochromatosis should receive age-appropriate screening for colorectal and breast cancers.5,35
Prognosis
- Patients with the clinical hereditary hemochromatosis phenotype have been shown to have higher in-hospital mortality (relative risk = 2.45; 95% CI, 2.27 to 2.64; 857 deaths) and out-of-hospital mortality (relative risk = 1.15; 95% CI, 1.00 to 1.33; 216 deaths) compared with control groups.16
- The presence of cirrhosis or a ferritin level greater than 2,000 ng per mL (2,000 mcg per L) at the time of diagnosis or in treated patients is associated with higher mortality, mostly liver-related deaths (standardized mortality ratio = 23.9; 95% Cl, 13.9 to 38.2).36
- Increased age, diabetes, alcohol consumption, and hepatic fibrosis are independent prognostic factors for death.36
- One study suggested that 20% of patients with hereditary hemochromatosis and cirrhosis will develop hepatocellular carcinoma.37
- Patients with diabetes have a twofold to sixfold increased risk of premature death if they have increased transferrin saturation or the HFE genotype.38
This article updates other articles on this topic by Crownover and Covey5 and Brandhagen, et al.39
Data Sources: A PubMed search was completed in Clinical Queries using the key term hereditary hemochromatosis. The search included meta-analyses, randomized clinical trials, clinical trials, and reviews. We also searched the Agency for Healthcare Research and Quality evidence reports, Clinical Evidence, the Cochrane database, Essential Evidence Plus, the Institute for Clinical Systems Improvement, and the evidence summary provided by AFP editors. Search dates: August 2020 and April 2021.
