Primary hyperparathyroidism is the most frequent cause of hypercalcemia in ambulatory patients. The condition is most common in postmenopausal women, although it can occur in persons of all ages, including pregnant women. If symptoms are present, they are attributable to hypercalcemia and may include weakness, easy fatigability, anorexia, or anxiety. However, most persons have no symptoms, and primary hyperparathyroidism usually is diagnosed after an elevated serum calcium level is found incidentally on multiphasic chemistry panel testing. Persistent hypercalcemia and an elevated serum parathyroid hormone level are the diagnostic criteria for primary hyperparathyroidism. Other causes of hypercalcemia are rare, and usually are associated with low (or sometimes normal) parathyroid hormone levels. Malignancy is the most frequent cause of hypercalcemia in hospitalized patients. Parathyroidectomy is the definitive treatment for primary hyperparathyroidism. When performed by experienced endocrine surgeons, the procedure has success rates of 90 to 95 percent and a low rate of complications. Asymptomatic patients who decline surgery and meet criteria for medical management must commit to conscientious long-term monitoring. Any unexplained elevation of the serum calcium level should be evaluated promptly to prevent complications from hypercalcemia.
Parerimary hyperparathyroidism is the most common cause of hypercalcemia in the outpatient setting.1 Most persons with this condition asymptomatic. However, recognition of primary hyperparathyroidism has increased dramatically since the introduction of multichannel autoanalyzers in the 1970s.2 The disorder can occur in persons of any age but is more common in persons older than 50 years.1 In the United States, its estimated incidence in persons older than 65 years is one case per 1,000 in men and two to three cases per 1,000 in women.1,3
Classic primary hyperparathyroidism with overt complications of osteitis fibrosa cystica, nephrolithiasis, and nephrocalcinosis is rare.1,2 Primary hyperparathyroidism usually is easily distinguishable from malignancy, which is the second most common cause of hypercalcemia. Laboratory measurements of the mediators of calcium metabolism are reliable and facilitate determination of etiologic factors in almost all patients with hypercalcemia.1
Parathyroid Glands
The four parathyroid glands normally are located behind the four poles of the thyroid gland. The glands secrete parathyroid hormone (PTH), which is the primary regulator of calcium homeostasis.4 The glands tightly regulate the extracellular calcium concentration within a narrow normal range.
The parathyroid glands respond within seconds to a low or falling serum calcium concentration (Figure 1). The chief cells of these glands can synthesize, process, and store PTH in a regulated manner and can replicate when chronically stimulated. These factors allow for short-, intermediate-, and long-term adaptability to fluctuations in the serum calcium level.4,5
PTH is an 84–amino-acid single-chain peptide that mobilizes calcium from the bones by osteoclastic stimulation. It also stimulates the kidneys to reabsorb calcium and convert 25-hydroxyvitamin D3 (produced in the liver) to the active form, 1,25-dihydroxyvitamin D3 which, in turn, stimulates gastrointestinal absorption of calcium. Low circulating concentrations of calcium stimulate PTH secretion, and high circulating concentrations of calcium depress PTH secretion. PTH is metabolized rapidly in the liver and kidneys; its circulating half-life is approximately two to five minutes.5
FIGURE 1.
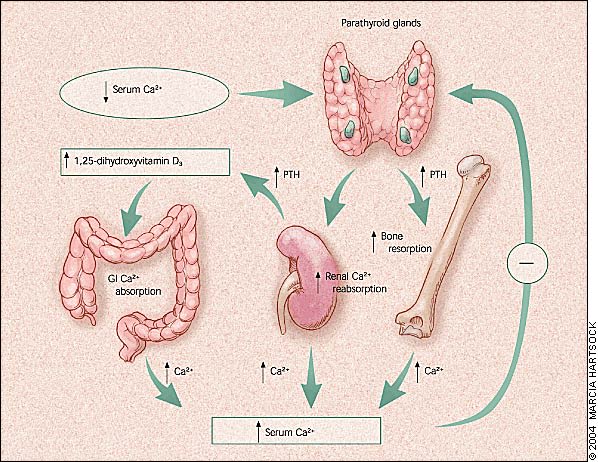
Parathyroid gland action. Low circulating serum calcium (Ca2+) concentrations stimulate the parathyroid glands to secrete parathyroid hormone (PTH), which mobilizes calcium from bones by osteoclastic stimulation. PTH also stimulates the kidneys to reabsorb calcium and to convert 25-hydroxyvitamin D3 (produced in the liver) to the active form, 1,25-dihydroxyvitamin D3, which stimulates gastrointestinal calcium absorption. High serum calcium concentrations have a negative feedback effect on PTH secretion. (GI = gastrointestinal)
Etiology and Pathogenesis
PRIMARY HYPERPARATHYROIDISM
Primary hyperparathyroidism is caused by the inappropriate secretion of PTH, which results in hypercalcemia. The condition usually occurs sporadically, although familial forms are well recognized.4
In 85 percent of patients with primary hyperparathyroidism, the underlying cause is an adenoma in a single parathyroid gland.1 Hypertrophy of all four parathyroid glands and multiple adenomas within the parathyroid glands account for the remainder of cases. Fewer than 0.5 percent of cases are caused by parathyroid malignancies.1
Most patients with primary hyperparathyroidism are postmenopausal women. The incidence of the condition increases with age, and the average age at diagnosis is 55 years. A small percentage of patients presents years after external neck irradiation. Lithium therapy also can be responsible for overactive parathyroid glands, with the excess activity persisting even after discontinuation of the drug.1
In 10 to 20 percent of patients, primary hyperparathyroidism is caused by an inherited hyperfunction of multiple parathyroid glands. These patients tend to be diagnosed at a younger age.1
Although rare, familial disorders should be considered in patients diagnosed with primary hyperparathyroidism. With several of these familial disorders, patients should be evaluated for significant associated abnormalities. Familial forms of primary hyperparathyroidism include multiple endocrine neoplasia type I and type II, neonatal severe primary hyperparathyroidism, hyperparathyroidism–jaw tumor syndrome, familial hypocalciuric hypercalcemia, and familial isolated hyperparathyroidism.6
Approximately 95 percent of patients with multiple endocrine neoplasia type I, or Werner's syndrome, have associated primary hyperparathyroidism.1,6,7 This syndrome is associated with various tumors, including pancreatic and pituitary adenomas.6,7 Patients with multiple endocrine neoplasia type II, or Sipple's syndrome, may develop a milder form of primary hyperparathyroidism.1,6,7 This syndrome is characterized primarily by medullary thyroid carcinoma and pheochromocytoma. Genetic testing for both syndromes is available in some medical centers.6,7
Neonatal severe primary hyperparathyroidism presents with severe hypercalcemia during the newborn period.1,6 Hyperparathyroidism–jaw tumor syndrome is a rare condition that usually presents in adolescents or young adults as a solitary adenoma associated with bone lesions in the jaw and with Wilms' tumor or renal cysts.6
Familial hypocalciuric hypercalcemia is caused by a single mutation in the calcium-sensing receptor, resulting in insensitivity to feedback inhibition of PTH secretion. In affected patients, the calcium/creatinine clearance ratio is less than 0.01, compared with a calcium/creatinine clearance ratio of greater than 0.02 in patients with primary hyperparathyroidism. Patients with familial hypocalciuric hypercalcemia are asymptomatic and require no treatment.1,6,8 Familial isolated hyperparathyroidism has no specific features. It is usually thought to be an expression of occult multiple endocrine neoplasia type I.6
SECONDARY AND TERTIARY HYPERPARATHYROIDISM
It is important to exclude both secondary and tertiary hyperparathyroidism, because the treatments are different. Secondary hyperparathyroidism is the result of a physiologic or pathophysiologic parathyroid response to hypocalcemia in an attempt to maintain calcium homeostasis. The condition can occur because of vitamin D deficiency or low calcium intake. The serum PTH level is elevated, and the serum calcium level may be normal or low, because of a diet that is limited in vitamin D or calcium, or because of deficiency secondary to malabsorption. In most instances, secondary hyperparathyroidism is caused by chronic renal failure, which results in a low concentration of 1,25-dihydroxyvitaminD3 because of decreased renal production.4,8
Tertiary hyperparathyroidism occurs because of prolonged hypocalcemia (usually secondary to chronic renal failure) that causes parathyroid gland hyperplasia. Autonomous oversecretion of PTH by the parathyroid glands results in hypercalcemia.4,8
Clinical Manifestations
A National Institutes of Health (NIH) consensus panel classified primary hyperparathyroidism into two categories: symptomatic and asymptomatic.9 [Evidence level C, expert opinion] Manifestations of symptomatic primary hyperparathyroidism secondary to hypercalcemia (which is becoming less common3) include overt bone disease; kidney stones; and nonspecific gastrointestinal, cardiovascular, and neuromuscular dysfunction.9
Asymptomatic primary hyperparathyroidism accounts for 75 to 80 percent of cases.10 Patients in this category have no signs or symptoms of hypercalcemia. Signs and symptoms that may be present but are not clearly associated with primary hyperparathyroidism include hypertension, left ventricular hypertrophy, valvular or myocardial calcification, peptic ulcer disease, pancreatitis, gout or pseudogout, normocytic normochromic anemia, weakness, easy fatigability, lassitude, anxiety, cognitive difficulties, somatic complaints, and clinical depression.1,2,4,9
Diagnosis
Persistent hypercalcemia and an elevated serum PTH level confirm the diagnosis of primary hyperparathyroidism (Figure 22–5,9,11). Further laboratory testing is unnecessary because other causes of hypercalcemia rarely are associated with elevated PTH levels. Radiologic testing cannot diagnose this disorder.1 The next most common cause of hypercalcemia is malignancy, which usually is associated with suppressed PTH levels.1,12
Hypercalcemia should be confirmed by repeated measurements of serum calcium concentrations, because all patients with primary hyperparathyroidism do not have demonstrable hypercalcemia every time the serum calcium level is measured.1,11 Preferably, tests should be performed with the patient fasting and with minimal venous occlusion. Use of thiazide diuretics should be discontinued two weeks before a serum calcium level measurement is repeated.
FIGURE 2. Diagnosis and Treatment of Primary Hyperparathyroidism
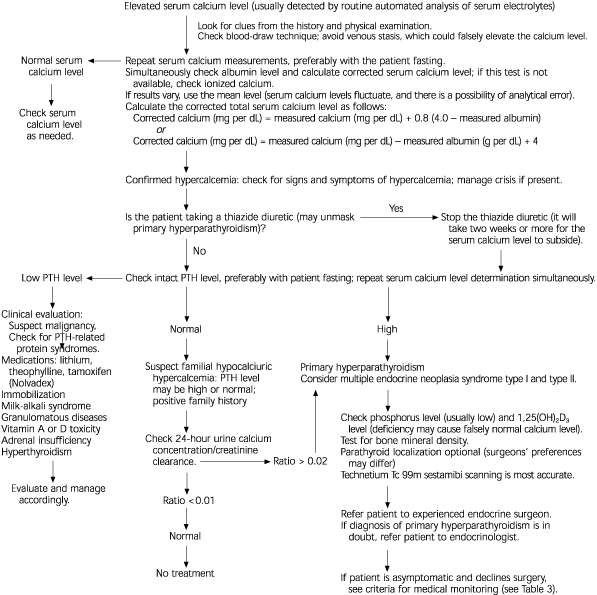
Algorithm for the diagnosis and treatment of primary hyperparathyroidism (PTH = parathyroid hormone; 1,25(OH)2D3 = 1,25-dihydroxyvitamin D3)
Information from references 2 through 5,9 and 11.
All serum calcium concentrations should be “corrected” to the prevailing serum albumin concentration. The total calcium level can be corrected for low albumin levels by adding 0.8 mg per dL (0.20 mmol per L) to the total serum calcium level for every 1.0 g per dL (10 g per L) by which the serum albumin concentration is lower than 4 g per dL (40 g per L).1 For example, in a patient with a total serum calcium level of 10 mg per dL (2.50 mmol per L) and an albumin level of 2.0 g per dL (20 g per L), the total calcium concentration is 11.6 mg per dL (2.90 mmol per L). Ionized calcium levels are unaffected by altered serum albumin levels. The NIH consensus panel9 did not recommend routine testing of the ionized calcium level for confirmation of hypercalcemia, because most physicians do not have access to a facility that can produce an accurate measurement.
Intact PTH (the entire 1 through 84 amino acid sequence) can be measured by immunoradiometric assay or immunochemical assay.4,5 Modern intact-PTH assays have no cross-reactivity with PTH-related protein. In approximately 10 percent of patients, the PTH level is not elevated but is in the upper one half of normal range.2 Vitamin D deficiency can cause false-normal calcium levels.5
Once the diagnosis of primary hyperparathyroidism is confirmed biochemically, bone mineral density should be measured in three sites (lumbar spine, hip, forearm), and the patient should be evaluated for renal complications.1,4,5,9 In women who are pregnant, fetal complications may include intrauterine growth retardation, low birth weight, preterm delivery, intrauterine fetal demise, postpartum neonatal tetany, and permanent hypoparathyroidism.3
Other possible causes of hypercalcemia must be considered in the differential diagnosis of primary hyperparathyroidism3,4,5,11 (Table 1).3
Treatment
SURGERY
Parathyroidectomy is the definitive treatment for primary hyperparathyroidism.1,2,3,9,13 When performed by experienced endocrine surgeons, this procedure has reported success rates of 90 to 95 percent with low complication rates.13 Parathyroidectomy should be offered to patients who are symptomatic and patients who meet the criteria for surgery established by the 2002 NIH consensus panel (Table 2).9 In pregnant women, it is preferable to perform parathyroidectomy after the first trimester.3,10
TABLE 2 Indications for Surgical Treatment of Primary Hyperparathyroidism
| Symptomatic primary hyperparathyroidism (nephrolithiasis, nephrocalcinosis, osteitis fibrosa cystica) | |
| Asymptomatic primary hyperparathyroidism | |
| Serum calcium level > 1.0 mg per dL (0.25 mmol per L) above the upper limits of normal | |
| Urinary calcium excretion > 400 mg per 24 hours (10 mmol per day) | |
| Creatinine clearance reduced by more than 30 percent compared with age-matched persons | |
| Bone density (lumbar spine, hip, or forearm) that is > 2.5 standard deviations below peak bone mass (T score -2.5) | |
| Patient age < 50 years, with asymptomatic primary hyperparathyroidism | |
| Medical surveillance not desirable or possible | |
| Surgery requested by patient | |
Information from reference 9.
The standard surgical approach is bilateral neck exploration, with identification of all four parathyroid glands.14 With the advent of preoperative localization studies, minimally invasive unilateral approaches have been developed.15–17 The procedure may be video-assisted, endoscopic, radio-guided, or image-guided unilateral exploration using local anesthesia.16–18 A unilateral approach may be used in the patient with a solitary adenoma that is preoperatively located with imaging studies.
Technetium Tc 99m sestamibi scanning is the most accurate technique (80 to 90 percent sensitivity) for localizing abnormal parathyroid glands.18 When this scanning is combined with single-photon emission computed tomography, the sensitivity is 91 percent, and the specificity is 98.8 percent.18–20 Other imaging modalities include ultrasonography, computed tomography, and magnetic resonance imaging.
An intraoperative rapid PTH test that can be performed in minutes is available to detect any remaining abnormal glands. If the intraoperative PTH level falls by more than 50 percent, surgery should be terminated.16–18,20 If the decline in the intraoperative PTH level is less than 50 percent, surgery should be extended, and full neck exploration may be necessary to look for other overactive glands.9 Ectopic parathyroid glands may be found in several locations (e.g., intrathyroidal, retroesophageal, mediastinal).
Immediate postoperative management focuses on establishing the success of the surgery and monitoring the patient for complications such as symptomatic hypocalcemia, bleeding, vocal cord paralysis, and laryngospasm. The serum calcium concentration typically reaches a nadir within 24 to 36 hours after surgery. The serum PTH level is in the normal range within 30 hours, although the secretory response to hypocalcemia may not return to normal for weeks. The patient should maintain a low-calcium diet until the serum calcium concentration is normal. Ampules of injectable calcium and other seizure precautions should be maintained at the bedside for emergency use.
Outpatient monitoring of serum and urinary calcium levels at frequent intervals may be necessary. After parathyroidectomy, patients with large adenomas may develop “hungry bone syndrome,” which is associated with hypocalcemia, hypophosphatemia, and low urinary calcium excretion.21 Persistent hypercalcemia and elevated intact PTH levels after surgery commonly are caused by an overlooked adenoma or incomplete resection of a hyperplastic gland. Less common causes are a failure to recognize and adequately treat parathyroid malignancy or an incorrect diagnosis of primary hyperparathyroidism.22
NONOPERATIVE INTERVENTION
Asymptomatic patients who meet the criteria suggested by the NIH guidelines may be candidates for medical monitoring rather than surgical intervention with parathyroidectomy9,23 (Table 3).9 [Evidence level C, consensus opinion] Commitment to conscientious, long-term medical monitoring at least semiannually is essential. Recommended surveillance includes biannual measurement of serum calcium levels, annual measurement of serum creatinine levels, and annual bone density testing.9
TABLE 3 Criteria for Medical Monitoring of Patients with Asymptomatic Primary Hyperparathyroidism
| Serum calcium level only mildly elevated |
| No previous episode of life-threatening hypercalcemia |
| Normal renal status (i.e., creatinine clearance of > 70 percent without nephrolithiasis or nephrocalcinosis) |
| Normal bone status (i.e., T score above -2.5 at lumbar spine, hip, and forearm) |
| Asymptomatic |
Information from reference 9.
There are no clinical factors that predict the prognosis of patients with asymptomatic hyperparathyroidism.9,23 Studies have demonstrated that up to 25 percent of asymptomatic patients develop indications for surgery during medical observation.1 Factors that can exacerbate hypercalcemia should be avoided and treated accordingly. Recommendations include modest intake of calcium (1,000 to 1,200 mg per day) and vitamin D (400 to 600 IU per day). Theoretically, low calcium intake could stimulate PTH production.1,13
Currently, no medical therapies are available to effectively cure primary hyperparathyroidism. In postmenopausal women, estrogen may reduce PTH-stimulated bone resorption. The effects of newer oral bisphosphonates, calcimimetics, and raloxifene are being studied.9,23 Medications may be tried in symptomatic patients who have severe concurrent illness but are poor surgical candidates. Bisphosphonates should be used with caution in patients with renal failure.
SECONDARY AND TERTIARY HYPERPARATHYROIDISM
Whenever possible, the underlying cause of secondary hyperparathyroidism should be removed. The goal of medical management is to normalize calcium levels. Supplementation of vitamin D and calcium is necessary. Patients with end-stage renal disease also need phosphate binders to decrease hyperphosphatemia. Parathyroidectomy may be necessary in patients who develop tertiary hyperparathyroidism and severe metabolic bone disease.4,5,24
