Sarcoidosis is a systemic granulomatous disease of unknown cause affecting young and middle-aged adults. Patients commonly present with bilateral hilar lymphadenopathy, pulmonary infiltrates, and ocular and skin lesions. The heart, liver, spleen, salivary glands, muscles, bones, kidneys, and central nervous system also may be involved. Diagnosis is based on clinicoradiologic findings plus histologic evidence of noncaseating epithelioid granulomas, and exclusion of other granulomatous diseases. Prognosis correlates with mode of onset, host characteristics, initial clinical course, and extent of disease. The optimal management of sarcoidosis has not been well defined. Although corticosteroids remain the mainstay of treatment, there is little evidence for the optimal initiation, dosage, or duration of therapy. Topical steroids may be considered for treatment of anterior uveitis and skin lesions. Systemic steroids are indicated for treatment of cardiac, nervous system, severe ocular, and symptomatic or progressive pulmonary involvement. There is little evidence for the efficacy of inhaled steroids. Cytotoxic agents and immunomodulators usually are reserved for treatment of complex or refractory disease. Of these agents, methotrexate is used more frequently because of its safety profile and possible steroid-sparing effects. Antimalarial agents are used frequently for skin lesions, and they have limited success in the treatment of pulmonary disease. Lung and cardiac transplantation is reserved for end-stage disease. Monitoring for symptoms of drug toxicity is essential, and prevention of osteoporosis must be addressed in patients taking long-term oral corticosteroids. It is not known if current therapy alters disease progression.
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that predominantly affects the lungs. The many forms and presentations of this disease and the lack of a single diagnostic test can make the diagnosis challenging. Physicians have limited guidance for the treatment of sarcoidosis because of a lack of high-quality randomized controlled trials (RCTs). This article reviews the epidemiology, etiology, clinical presentation, diagnosis, and current evidence on the treatment of pulmonary and extrapulmonary sarcoidosis.
Strength of Recommendations
| Key clinical recommendation | Labels | References |
|---|---|---|
| Treatment is not indicated for patients with stage i sarcoidosis alone. | A | 5 |
| Treatment of patients with stage II or III sarcoidosis with oral steroids for six to 24 months improves chest radiograph findings during therapy. | A | 5 |
| For pulmonary sarcoidosis, the initiation dosage is 20 to 40 mg per day of prednisone or its equivalent for one to three months. Every-otherday dosing also may be considered. In patients who respond, the prednisone dose should be tapered to 5 to 10 mg per day or every other day for a minimum of 12 months. | C | 1 |
| Inhaled corticosteroids may be considered for the symptomatic relief of cough, although there are few data supporting their use. | A | 1,5 |
| Calcitonin (Miacalcin) and bisphosphonates may be useful in preventing osteoporosis in patients with sarcoidosis taking long-term steroid therapy. | B | 8,9 |
| Calcium and vitamin D supplementation should be used with caution because of the risk of hypercalcemia. | C | 1 |
| Cytotoxic agents and immunomodulators may be considered for patients with complicated or severe refractory sarcoidosis, or as steroid-sparing agents. | B | 15–30,34,38 |
Epidemiology
Sarcoidosis affects men and women of all races and ages. The condition usually presents in adults younger than 40 years, most frequently between 20 and 29 years of age. It is slightly more predominant in women than in men, with an incidence of 6.3 and 5.9 cases per 100,000 person-years, respectively. The lifetime risk of sarcoidosis for U.S. whites is estimated at 0.85 percent compared with 2.4 percent in U.S. blacks. Sarcoidosis is most prevalent in Swedes, Danes, and U.S. blacks. Mortality from sarcoidosis, usually caused by respiratory failure, approaches 1 to 5 percent.1,2 Because most patients with sarcoidosis do not die of the disease, the medical challenge is to help them live well with their symptoms.
Etiology and Pathogenesis
Multiple causes of sarcoidosis have been proposed. Evidence exists to support genetic inheritance, infectious transmission, and shared exposure to environmental agents.3 Infectious organisms such as viruses, mycobacteria, Borrelia burgdorferi, and Propionibacterium acnes have been implicated as potential causes of sarcoidosis.1 Environmental exposure to beryllium, aluminum, and zirconium can result in a granulomatous response similar to that of sarcoidosis.1 Current theory suggests that disease develops in genetically predetermined hosts who are exposed to certain environmental agents that trigger an exaggerated inflammatory immune response leading to granuloma formation.1,2
The characteristic lesion of sarcoidosis is a discrete, noncaseating, epithelioid granuloma. The early sarcoid reaction occurs when activated T cells and macrophages accumulate at sites of ongoing inflammation. These activated cells release chemoattractants and growth factors that result in cellular proliferation and granuloma formation. Sarcoid granulomas resolve or leave behind fibrotic changes. The factors leading to fibrosis are poorly understood.1
Clinical Presentation and Organ Involvement
The clinical presentation of sarcoidosis depends on ethnicity, duration of illness, site and extent of organ involvement, and activity of the granulomatous process. The usual modes of presentation include non-specific constitutional symptoms or symptoms related to organ-specific involvement.2 Thirty to 50 percent of patients are asymptomatic and are diagnosed on routine chest radiographs. One third of patients have non-specific symptoms of fever, fatigue, weight loss, and malaise. This presentation is more common in blacks and Asian Indians.1
Acute sarcoidosis is more common in whites than in blacks and usually is associated with spontaneous remission within two years. Spontaneous remission also occurs in patients with Löfgren’s syndrome, which consists of bilateral hilar lymphadenopathy, ankle arthritis, erythema nodosum, fever, myalgia, and weight loss.1 Chronic sarcoidosis presents insidiously with symptoms related to the organ involved, such as cough and dyspnea from pulmonary infiltration. Chronic sarcoidosis commonly follows a relapsing and protracted time course.2 Chronic progressive disease affects 10 to 30 percent of patients. Spontaneous remission occurs in approximately two thirds of these patients. Blacks have increased rates of pulmonary involvement, a worse long-term prognosis, and more frequent relapses.1
The lungs are involved in more than 90 percent of patients, with sarcoid usually presenting as interstitial disease. Symptoms are dry cough, dyspnea, and chest discomfort. Pulmonary sarcoidosis has an unpredictable course that may result in spontaneous remission or lead to progressive loss of lung function with fibrosis. There are four stages of pulmonary sarcoidosis (Table 12 and Figure 1). Patients with stage I or II disease may have no symptoms, whereas stages III and IV can be characterized by progressive dyspnea, loss of lung function, and fibrosis. Airway involvement can occur and may result in airflow limitation, persistent cough and, in severe cases, bronchiectasis. Spontaneous remission can be expected in 55 to 90 percent of patients with stage I disease, 40 to 70 percent with stage II disease, 10 to 20 percent with stage III disease, and zero to 5 percent with stage IV disease.1,2
TABLE 1 Stages of Sarcoidosis
| Stage | Chest radiograph characteristics | Frequency (%) | Rates of spontaneous remission (%) |
|---|---|---|---|
| 0 | Normal | 5 to 10 | — |
| I | BHL | 50 | 55 to 90 |
| II | BHL and parenchymal infiltrates | 25 | 40 to 70 |
| III | Parenchymal infiltrates without BHL | 15 | 10 to 20 |
| IV | Signs of fibrosis | 5 to 10 | 0 to 5 |
BHL = bilateral hilar lymphadenopathy.
Adapted with permission from Costabel U. Sarcoidosis: clinical update. Eur Respir J Suppl 2001;32:59s,62s,63s.
Figure 1
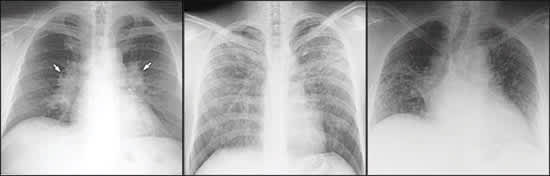
Posteroanterior views of the chest showing (left) bilateral hilar adenopathy, indicative of stage I sarcoidosis, and (center) stage III sarcoidosis, and (right) single view of the chest showing stage IV sarcoidosis.
Sarcoidosis can involve multiple organs and cause a variety of clinical symptoms (Table 2).1,2 Although granulomas are found in 50 to 80 percent of liver biopsies, they rarely result in clinically significant hepatic dysfunction and usually cause only mild abnormalities in liver function test results.
TABLE 2 Organ Involvement in Sarcoidosis
| Organ | Patients (%) |
|---|---|
| Mediastinal lymph nodes | 95 to 98 |
| Lungs (cough, chest pain, airflow limitation, bronchiectasis, progressive dyspnea, fibrosis) | > 90 |
| Liver (mild increase of hepatic enzymes usual, hepatic failure rare) | 50 to 80 |
| Spleen (splenomegaly, usually asymptomatic) | 40 to 80 |
| Eyes (uveitis common, may lead to blindness) | 20 to 50 |
| Musculoskeletal system (joint pain usual, myopathy and bone cysts rare) | 25 to 39 |
| Peripheral lymph nodes (palpable, discrete, mobile, nontender; usually cervical, axillary, epitrochlear, and inguinal) | 30 |
| Hematologic (anemia, leukopenia) | 4 to 40 |
| Skin (nonspecific lesions, erythema nodosum, lupus pernio) | 25 |
| Nervous system (cranial nerve palsies, space-occupying lesions, peripheral neuropathy, diabetes insipidus with pituitary involvement) | 10 |
| Heart (conduction abnormalities, infiltrative cardiomyopathy, sudden death) | 5 |
| Hypercalcemia (may cause nephrocalcinosis, renal stones, and renal failure) | 2 to 10 |
| Parotid glands (unilateral or bilateral parotitis) | < 6 |
| Gastrointestinal (usually stomach, also esophagus, appendix, rectum, pancreas) | < 1 |
| Kidneys (interstitial nephritis, nephrocalcinosis) | Rare |
Adapted with permission from Costabel U. Sarcoidosis: clinical update. Eur Respir J Suppl 2001;32:59s, with additional information from reference 1.
Twenty-five percent of patients have skin involvement. Lesions can range from non-specific maculopapular eruptions, such as plaques and nodules, to erythema nodosum and lupus pernio. Erythema nodosum is the characteristic lesion of acute sarcoid, which has a good prognosis. Lupus pernio consists of indurated plaques and discoloration of the nose, cheeks, lips, and ears and usually indicates a chronic disease course that is unlikely to result in spontaneous remission. Adverse prognostic factors in patients with sarcoidosis are summarized in Table 3.1,2
TABLE 3 Adverse Prognostic Factors in Patients with Sarcoidosis
Children with sarcoidosis have the same organ involvement as adults but a more favorable prognosis. Sarcoidosis rarely interferes with pregnancy, but the disease can worsen at six months’ postpartum. New diagnoses of sarcoidosis in the elderly are rare and should be distinguished from a local sarcoid reaction that occurs with malignancies.1
Diagnosis
Because of its nonspecific presentation, the diagnosis of sarcoidosis can be challenging. It has been shown recently that the diagnosis often is delayed.4 Patients with pulmonary symptoms have the more prolonged diagnosis. Those with dermatologic findings are diagnosed with less delay.4
The essential factors for diagnosis include compatible clinicoradiologic features, histologic proof of noncaseating epithelioid granulomas, and exclusion of similar diseases.1,2 Posteroanterior chest radiographs are useful in staging the disease. Transbronchial lung biopsy is recommended in most cases, but easily accessible skin lesions or peripheral lymph nodes also may be sampled.1
When biopsy is refused or is negative, certain biologic markers may assist in making a diagnosis. Bronchoalveolar lavage fluid with a CD4 to CD8 ratio greater than 3.5, panda and lambda patterns on gallium scan, or an angiotensin-converting enzyme (ACE) level double the normal value may provide additional diagnostic information, although each factor lacks specificity.1,2 ACE levels usually are followed as a marker of disease activity rather than for diagnostic purposes. Other potential causes of granuloma formation are listed in Table 4.1
The initial evaluation of patients with sarcoidosis aims to confirm the diagnosis, assess the extent and severity of involvement, identify stable versus progressive disease, and judge whether therapy will benefit the patient (Table 5).1 Follow-up at recommended intervals (Table 6)1 and consultation with subspecialists likely will be necessary, depending on disease manifestation.
TABLE 5 Recommended Initial Evaluation of Patients with Sarcoidosis
| History (occupational and environmental exposure, symptoms) |
| Physical examination |
| Posteroanterior chest radiograph |
| Pulmonary function tests: spirometry and diffusing capacity of the lung for carbon dioxide |
| Peripheral blood counts: white blood cells, red blood cells, platelets |
| Serum chemistries: calcium, liver enzymes (alanine transaminase, aspartate transaminase, alkaline phosphatase), creatinine, blood urea nitrogen |
| Urine analysis |
| Electrocardiograph |
| Routine ophthalmologic examination |
| Tuberculin skin test |
Adapted with permission from Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med 1999;160:744.
TABLE 6 Surveillance Intervals for Patients with Sarcoidosis*
| Stage I: initially every six months, then annually if stable |
| Stage II, III, IV: initially every three to six months, monitor indefinitely |
| Serious extrapulmonary involvement: monitor indefinitely |
| Monitor three years after cessation of therapy for potential relapse, subsequent follow-up not necessary if stable |
*—Follow-up should include symptom review, physical examination, chest radiograph, spirometry; further testing depends on organ involvement.
Information from reference 1.
Treatment
PULMONARY THERAPY
Systemic Corticosteroids. Oral corticosteroids are the mainstay of treatment for pulmonary sarcoidosis. In a Cochrane review5 of corticosteroids for pulmonary sarcoidosis, treatment with oral steroids for six to 24 months improved chest radiograph findings compared with placebo. Patients with interstitial lung disease (stages II and III) had benefits in global scores and chest radiographs.5 Data show that no treatment is necessary for patients with stage I disease (bilateral hilar lymphadenopathy alone).5,6
It is not known whether steroids improve long-term lung function or favorably alter disease progression.5 A recent meta-analysis of mortality of intrathoracic sarcoidosis raises the possibility that a lower threshold for administering corticosteroids is associated with an increased mortality risk in referral centers.7 Few studies have looked at long-term follow-up after therapy with oral steroids. One recent RCT that examined follow-up after therapy with oral and inhaled steroids concluded that immediate treatment of stages II and III sarcoidosis, but not stage I, improved the five-year prognosis with regard to lung-function variables and the need for further treatment.5
There are no specific recommendations about when to initiate corticosteroid treatment. Data suggest that oral steroids should be used in patients with stages II and III disease, with moderate to severe or progressive symptoms and chest radiograph changes. It is unclear if asymptomatic patients will ever need therapy, even if they have diffuse lung infiltration. Because there is no consensus on the optimal dosage or duration of therapy, the course is individualized for each patient. A recent joint statement of the American Thoracic Society, the European Respiratory Society, and the World Association of Sarcoidosis and Other Granulomatous Disorders1 included the following guidelines: an initiation dose of prednisone of 20 to 40 mg per day or its equivalent is recommended. Every-otherday dosing may be considered. Patients should be evaluated after one to three months for response. Patients who fail treatment after three months usually will not respond to a more protracted course of treatment. In responders, the prednisone dosage should be tapered to 5 to 10 mg per day or to an every-otherday regimen, and therapy should continue for a minimum of 12 months.1 There is no consensus guidance on treatment beyond two years.2 Patients must be monitored after cessation of treatment for possible relapse; some patients will require long-term low-dose therapy to prevent recurrent disease.1
The risk of osteoporosis must be addressed in patients taking prolonged systemic corticosteroids. Although few studies have addressed this issue, the bisphosphonate alendronate (Fosamax) and nasal calcitonin (Miacalcin) have been shown to prevent osteoporosis in patients with sarcoidosis.8,9 Although not available in the United States, deflazacort, a prednisolone derivative with bone-sparing effects, was favored in one study over prednisone in the treatment of chronic sarcoidosis.10 Calcium and vitamin D supplementation should be used with caution because of the risk of hypercalcemia and hypercalciuria in patients with sarcoidosis.1
Inhaled Corticosteroids. There is little evidence for the efficacy of inhaled steroids in the treatment of pulmonary sarcoidosis.5 Theoretically, inhaled steroids should provide relief for the endobronchial component of sarcoidosis (i.e., cough). Studies have reported conflicting results about the efficacy of inhaled budesonide (Rhinocort) on disease stages I, II, and III.11–13 One study13 showed improvement in symptoms and inspiratory vital capacity with inhaled budesonide, but no change in chest radiograph findings, ACE level, or other lung function parameters. A recent RCT14 of inhaled fluticasone (Flovent) in the treatment of chronic stable sarcoidosis showed no benefit in lung function and chest radiographs, but a positive tendency toward symptomatic relief that was not statistically significant. Inhaled corticosteroids may be considered for the symptomatic relief of cough, although limited data support their use.1,5
Cytotoxic Agents. Cytotoxic agents may benefit patients who do not respond to corticosteroids. They are usually combined with oral steroids but sometimes used as single agents. Limited evidence exists to guide physicians in the initiation, dosage, and duration of therapy. Because of safety factors, methotrexate (Rheumatrex) and azathioprine (Imuran) are the preferred agents.1,2
Clinical data on the effects of methotrexate in the treatment of pulmonary sarcoidosis are limited to a few series and some case reports. A recent small RCT15 showed that 12 months of therapy with methotrexate was beneficial in patients with acute, symptomatic sarcoidosis. Other trials using methotrexate at 10 to 25 mg per week show its possible benefit as a steroid-sparing agent. Relapses were frequent after discontinuation of treatment, suggesting that methotrexate suppresses but does not cure the disease.1,15,16 Hypersensitivity pneumonitis and hepatotoxicity have been described in patients treated with methotrexate. This drug should be avoided in patients with renal failure.1 Folate given concomitantly decreases toxicity.
Azathioprine has been shown to be effective in a limited number of cases.2 Small clinical trials have demonstrated that azathioprine, when used for treatment of chronic pulmonary sarcoidosis, is effective as a corticosteroid-sparing agent.17,18 An increased risk of malignancy has been reported in transplant patients taking azathioprine.1
Chlorambucil (Leukeran) has been used as an alternative to steroids in a limited number of patients.19,20 Because of its increased risk of malignancy, this therapy has been abandoned by most physicians.1 Little is described about the use of cyclophosphamide (Cytoxan) in pulmonary sarcoidosis. High toxicity limits its use to patients with severe refractory disease.1
Immunomodulators. Immunomodulators have been used successfully in select patients with pulmonary sarcoidosis. The action of these drugs stems from immunosuppressive characteristics. Many of them modulate the activity of tumor necrosis factor alpha (TNF-α). TNF-α secreted by macrophages has been implicated in granuloma formation.21
In one study,22 the antimalarial agent chloroquine (Aralen) has been shown to decrease disease activity in advanced pulmonary sarcoidosis. Case reports23 also have suggested benefit in pulmonary sarcoidosis with the use of hydroxychloroquine (Plaquenil). These agents are more likely to be suppressive than curative and may work by inhibiting macrophage production of TNF-α.22 Because of the potential for retinal toxicity, patients on this regimen must have frequent ophthalmologic examinations.1 Despite a theoretic benefit on T helper-cell suppression, cyclosporine (Neoral) at conventional dosages has not been beneficial in the treatment of pulmonary sarcoidosis.24
Pentoxifylline (Trental) inhibits TNF-α production from alveolar macrophages in patients with sarcoidosis.25 In one small clinical trial, pentoxifylline was promising as a steroid-sparing agent in progressive pulmonary sarcoidosis.26 Through its anti–TNF-α activity, infliximab (Remicade) has presumptive usefulness in sarcoidosis treatment. Data limited to case reports show promising results in the treatment of complicated or refractory pulmonary sarcoidosis.27,28 Thalidomide (Thalomid) has been shown to be effective on certain dermatologic lesions, yet its action in pulmonary sarcoidosis is less well described.29 Case reports show some positive response in patients unable to tolerate corticosteroids.30 Thalidomide’s mechanism of action also derives from anti–TNF-α activity.31
A recent Cochrane review of immunosuppressive and cytotoxic therapy for pulmonary sarcoidosis was unable to recommend the use of these agents.32 High-quality studies are needed to examine the use of these drugs, especially the newer immunomodulators that act on TNF-α, for treatment of pulmonary sarcoidosis.
Surgical Intervention. Life-threatening hemoptysis may occur in advanced fibro-cystic sarcoidosis with bronchiectasis and mycetomas. Surgical resection and embolization of bronchial arteries may control the bleeding. Hemoptysis also may respond to conservative management with antibiotics, corticosteroids, bed rest, and cough suppression.2 End-stage pulmonary sarcoidosis with cor pulmonale may warrant supplemental oxygen, diuretics, and bronchodilators for airway obstruction.2 Lung transplantation has been performed successfully in patients with end-stage sarcoidosis. Granuloma formation has been described in lung allografts, although it usually is not clinically relevant.33
Therapy for Extrapulmonary Sarcoidosis
Despite the lack of RCTs demonstrating therapeutic efficacy, corticosteroids are still the mainstay of treatment for extrapulmonary sarcoidosis. Combination or adjuvant therapy has been investigated in many small studies.
OPHTHALMOLOGIC SARCOIDOSIS
Acute anterior uveitis usually clears spontaneously or after treatment with topical corticosteroids.1,2 Systemic corticosteroids are indicated for treatment of optic neuritis and uveitis resistant to topical agents.2,34 If ophthalmic surgery is necessary, it should be delayed until the ocular condition is inactive.34 One study showed methotrexate as an effective adjunctive agent in the treatment of chronic sarcoid-associated panuveitis.35
CUTANEOUS SARCOIDOSIS
Erythema nodosum, presenting alone or as part of Löfgren’s syndrome, usually resolves in six to eight weeks with nonsteroidal anti-inflammatory drugs alone.1 Systemic corticosteroids are used to treat lupus pernio, which indicates chronic sarcoidosis and a poor prognosis.2
In addition to intralesional steroid injections and topical corticosteroids with occlusive dressings,3 one study of 12 patients showed minocycline hydrochloride (Dynacin) and doxycycline (Vibramycin) to be effective.36 Systemic corticosteroids for large or disfiguring lesions and adjunctive therapy with methotrexate and hydroxychloroquine also have been shown to be beneficial.3 Thalidomide may be well tolerated and useful in the treatment of chronic cutaneous sarcoidosis.29
NEUROSARCOIDOSIS
Cranial neuropathy, persistent peripheral neuropathy, and intracranial lesions can be treated medically with oral corticosteroids (40 to 80 mg per day).3 Surgical intervention is necessary for hydrocephalus and symptomatic mass lesions.3 Adjunctive therapy with cyclosporine and azathioprine may be useful, especially in patients with cortisone-refractory neurosarcoidosis.37,38
CARDIAC SARCOIDOSIS
Treatment of cardiac sarcoidosis with corticosteroids is likely to be beneficial but is not well described in the literature. Surgical options include placement of pacemakers or cardioverterdefibrillators, resection of affected areas of the heart, and cardiac transplantation.3,39 Pharmacologic therapy for treatment of congestive heart failure and arrhythmias should be used when indicated.39
Drugs for the treatment of sarcoidosis are listed in Table 7.40 The primary care physician should consult subspecialists before prescribing cytotoxic or immunomodulator agents.
TABLE 7 Drugs Commonly Used for Treatment of Sarcoidosis
| Drug | Usual dosage | Common toxicities | Comments | |
|---|---|---|---|---|
| Corticosteroids (prednisone, prednisolone) | Initial: 20 to 40 mg daily Maintenance: 5 to 10 mg daily or every other day | Diabetes, hypertension, osteoporosis, insomnia, increased risk of infection | Most widely studied drug for sarcoidosis | |
| Antimalarial agents | ||||
| Chloroquine (Aralen) | 500 mg daily | Nausea | Useful for skin disease and hypercalcemia | |
| Hydroxychloroquine (Plaquenil) | 200 to 400 mg per day | Ocular toxicity | — | |
| Methotrexate (Rheumatrex) | 10 to 20 mg per week; dosage adjusted for toxicity | Nausea, neutropenia, hepatotoxicity, pulmonary fibrosis (rare) | Useful for chronic sarcoidosis; takes up to six months to become effective | |
| Azathioprine (Imuran) | 50 to 150 mg daily; dosage adjusted for toxicity | Nausea, neutropenia | Not as widely studied as methotrexate; neutropenia and nausea more common problems with this agent than with methotrexate | |
| Pentoxifylline (Trental) | 400 to 1,200 mg daily; dosage adjusted for toxicity | Nausea | Reports limited to use for acute disease | |
| Thalidomide (Thalomid) | 50 to 200 mg daily; dosage adjusted for toxicity | Somnolence, teratogenicity (major concern), constipation, peripheral neuropathy | Most useful for skin disease; not as effective for pulmonary disease | |
| Cyclophosphamide (Cytoxan) | Oral: 50 to 150 mg per day IV pulse: 500 to 1,500 mg every two to four weeks; dosage adjusted for toxicity | Neutropenia, nausea, cystitis, carcinogenicity | Effective, but toxicity limits its use to refractory cases | |
| Cyclosporine (Neoral) | 25 to 200 mg daily; dosage adjusted for toxicity (blood values used to monitor for toxicity) | Increased risk of infection, renal failure, hypertension, carcinogenicity | Highly variable reports on effectiveness; may be useful for treatment of neurosarcoidosis | |
| Infliximab (Remicade) | 5 mg per kg IV initially, week two, and then every four to eight weeks | Increased rate of infection, especially tuberculosis; allergic reaction to infusion; cannot give to patients with congestive heart failure; possible carcinogenicity | Few data about effectiveness, little information about dosage | |
IV = intravenous.
Adapted with permission from Baughman RP, Lower EE, du Bois RM. Sarcoidosis. Lancet 2003;361:1115.
