Inborn errors of metabolism (IEM), although individually rare, occur in 1 out of every 1,500 births. The first opportunity to detect IEM occurs during preconception counseling, when pregnant women and couples considering future pregnancies can undergo carrier screening. For individuals of all ethnic backgrounds, the screening includes testing for a variety of IEM and non-IEM. For individuals of Ashkenazi Jewish descent, carrier screening, per the American College of Medical Genetics and Genomics, also includes testing for Tay-Sachs disease and four other IEM. Inborn errors of metabolism can present in utero; in newborns; or in children, adolescents, and adults. Some IEM can be detected in utero with the use of ultrasonography. Most commonly, IEM are detected at newborn screening. Expanded newborn screening, which now includes 34 core conditions, allows for diagnosis in the newborn period and provides the opportunity for early institution of available treatments. However, some newborns present with symptoms consistent with an IEM before the availability of pending newborn screening results or present with symptoms attributable to an IEM not detectable with screening. Such situations are medical emergencies requiring immediate consultation with a metabolic specialist. If a delay occurs in obtaining consultation, initial treatment involves discontinuing feeding and providing high-rate glucose infusions. Some IEM present later in life. Children may develop and present with dysmorphic facial features. In some cases, symptoms may not appear until adolescence or adulthood when patients have residual enzyme activity that allows for slow accumulation of toxic molecules over time. Long-term treatments are effective for some IEM. Treatments include dietary restrictions and enzyme-replacement therapies.
Inborn errors of metabolism (IEM) are genetic conditions that block metabolic pathways involved in the breakdown of nutrients and the generation of energy. Perturbation of these metabolic pathways results in a spectrum of clinical findings affecting multiple organ systems. The diagnosis of IEM is challenging because the clinical presentation is often nonspecific; however, more IEM are now included in recommended newborn screening, which helps for early diagnosis. Therefore, knowledge of IEM has become essential for physicians. Although individual IEM are rare, the combined incidence is 1 out of every 1,500 births.1 This review discusses IEM disorders from preconception to adulthood.
WHAT IS NEW ON THIS TOPIC
The American College of Obstetricians and Gynecologists has classified expanded carrier screening as an acceptable pre-pregnancy and prenatal screening strategy for all patients. Expanded screening refers to concurrently screening for as many as several hundred conditions, including both IEM and non-IEM.
The initial treatment for all newborns and children with a suspected IEM comprises ending the buildup of toxic metabolites by discontinuing feeds and by preventing catabolism by giving glucose at a high infusion rate (5 to 10 g per kg per hour).
SORT: KEY RECOMMENDATIONS FOR PRACTICE
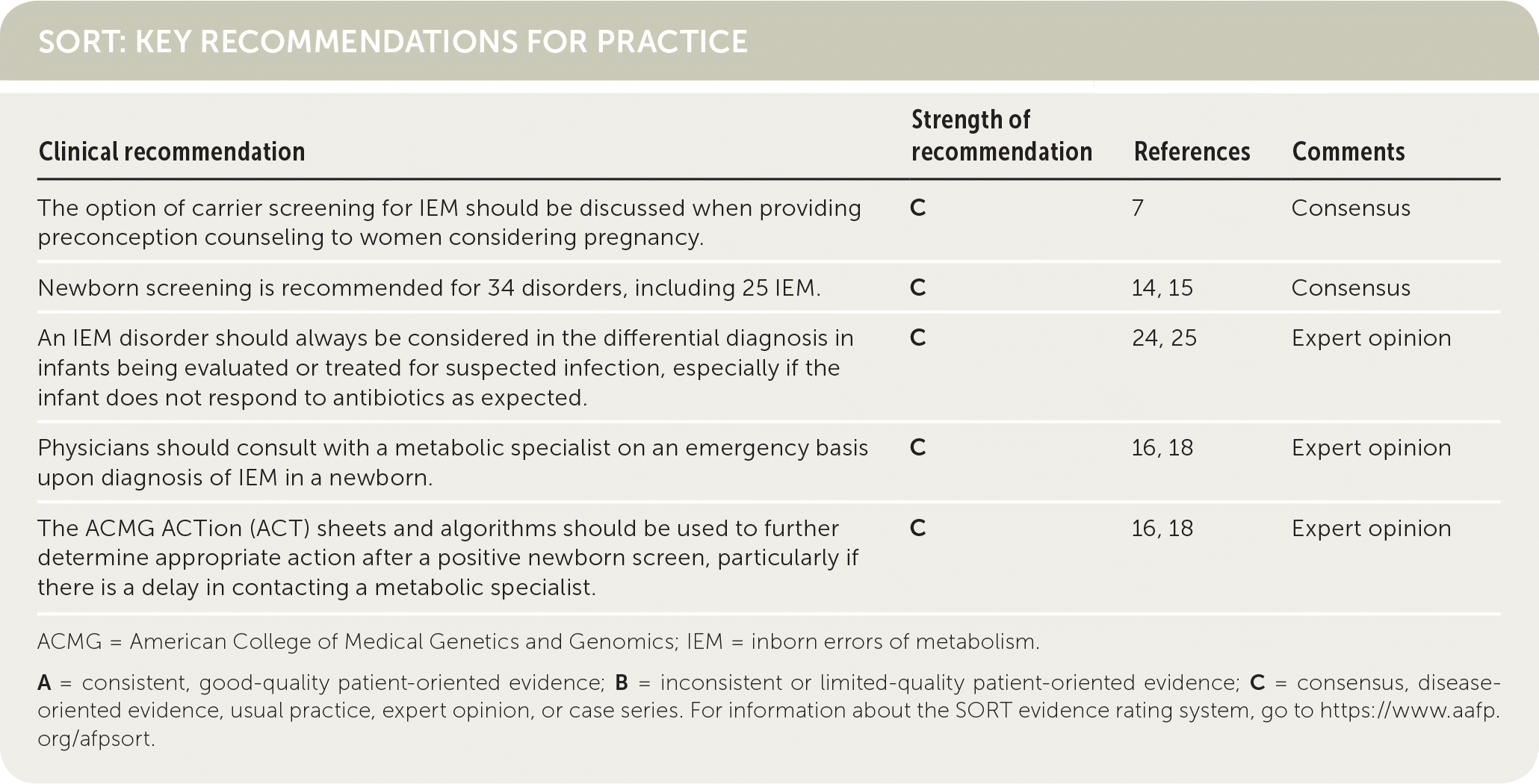
| Clinical recommendation | Strength of recommendation | References | Comments |
|---|---|---|---|
| The option of carrier screening for IEM should be discussed when providing preconception counseling to women considering pregnancy. | C | 7 | Consensus |
| Newborn screening is recommended for 34 disorders, including 25 IEM. | C | 14, 15 | Consensus |
| An IEM disorder should always be considered in the differential diagnosis in infants being evaluated or treated for suspected infection, especially if the infant does not respond to antibiotics as expected. | C | 24, 25 | Expert opinion |
| Physicians should consult with a metabolic specialist on an emergency basis upon diagnosis of IEM in a newborn. | C | 16, 18 | Expert opinion |
| The ACMG ACTion (ACT) sheets and algorithms should be used to further determine appropriate action after a positive newborn screen, particularly if there is a delay in contacting a metabolic specialist. | C | 16, 18 | Expert opinion |
ACMG = American College of Medical Genetics and Genomics; IEM = inborn errors of metabolism.
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.
Screening
PRECONCEPTION
The first opportunity to address IEM occurs with testing of asymptomatic future parents. Certain populations have increased carrier rates for IEM, and preconception screening has been shown to decrease disease prevalence.
Carrier testing first began in the Ashkenazi (Eastern European) Jewish population in the early 1970s with preconception screening for carriers of Tay-Sachs disease.2,3 With the advent of carrier screening, the incidence of Tay-Sachs disease decreased by 90% between 1970 and 1993 in the Jewish populations of North America.3 The American College of Obstetricians and Gynecologists (ACOG) and the American College of Medical Genetics and Genomics (ACMG) have expanded the list of recommended carrier testing for IEM beyond Tay-Sachs disease in the Ashkenazi Jewish population (Table 1).4–6
TABLE 1. IEM and Non-IEM Disorder Carrier Screening Recommendations for Adults of Ashkenazi Jewish Descent from the ACMG and the ACOG
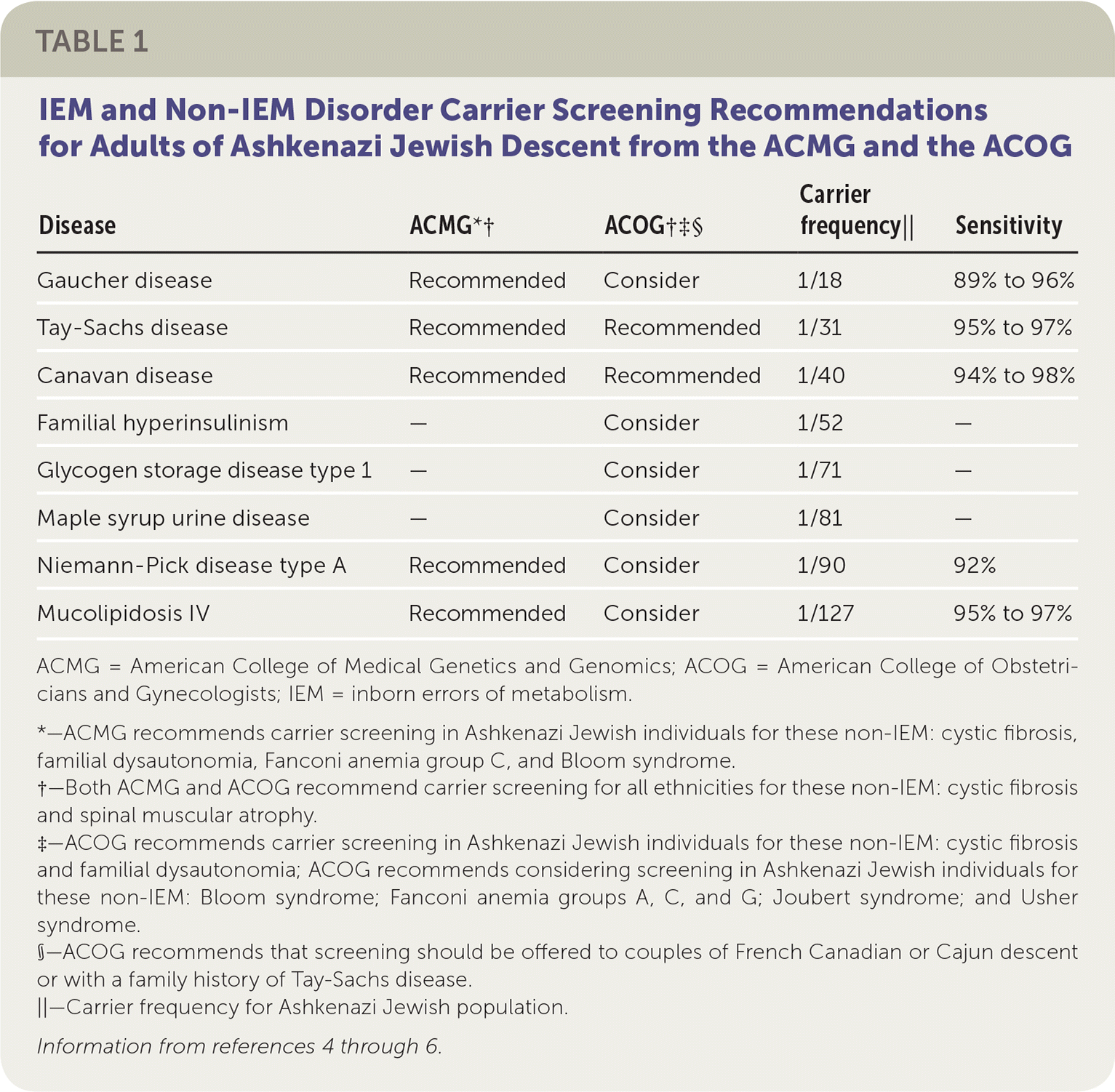
| Disease | ACMG*† | ACOG†‡§ | Carrier frequency|| | Sensitivity |
|---|---|---|---|---|
| Gaucher disease | Recommended | Consider | 1/18 | 89% to 96% |
| Tay-Sachs disease | Recommended | Recommended | 1/31 | 95% to 97% |
| Canavan disease | Recommended | Recommended | 1/40 | 94% to 98% |
| Familial hyperinsulinism | — | Consider | 1/52 | — |
| Glycogen storage disease type 1 | — | Consider | 1/71 | — |
| Maple syrup urine disease | — | Consider | 1/81 | — |
| Niemann-Pick disease type A | Recommended | Consider | 1/90 | 92% |
| Mucolipidosis IV | Recommended | Consider | 1/127 | 95% to 97% |
ACMG = American College of Medical Genetics and Genomics; ACOG = American College of Obstetricians and Gynecologists; IEM = inborn errors of metabolism.
*—ACMG recommends carrier screening in Ashkenazi Jewish individuals for these non-IEM: cystic fibrosis, familial dysautonomia, Fanconi anemia group C, and Bloom syndrome.
†—Both ACMG and ACOG recommend carrier screening for all ethnicities for these non-IEM: cystic fibrosis and spinal muscular atrophy.
‡—ACOG recommends carrier screening in Ashkenazi Jewish individuals for these non-IEM: cystic fibrosis and familial dysautonomia; ACOG recommends considering screening in Ashkenazi Jewish individuals for these non-IEM: Bloom syndrome; Fanconi anemia groups A, C, and G; Joubert syndrome; and Usher syndrome.
§—ACOG recommends that screening should be offered to couples of French Canadian or Cajun descent or with a family history of Tay-Sachs disease.
||—Carrier frequency for Ashkenazi Jewish population.
In addition, ACOG has recently classified expanded carrier screening as an acceptable pre-pregnancy and prenatal screening strategy for all patients.7 Expanded screening refers to concurrent screening for as many as several hundred conditions—both IEM and non-IEM—and is more accessible because of the decreased cost of DNA analysis.
Because of the previous recommendations and decreased testing costs, physicians in family medicine, obstetrics, and maternal fetal medicine are now able to offer preconception screening for inherited disorders; however, preconception screening detects only the most common DNA variants associated with a particular IEM disorder. Thus, a normal screening test does not always eliminate the possibility of an IEM disorder.
NEWBORN
IEM disorder screening began in the 1960s with Dr. Robert Guthrie's development of a screening test for phenylketonuria (PKU) from a blood spot8; early knowledge of the disorder allowed for treatment of PKU with diet restriction of the amino acid phenylalanine.9 Expanded newborn screening beyond PKU has occurred largely as a result of the introduction of tandem mass spectrometry, which allows for testing of multiple metabolic conditions from a single blood spot.10,11
Sensitivity for newborn screening using tandem mass spectrometry is 99.9%, and specificities range from 99.9% to 99.99%.12,13 Because of the low prevalence of these conditions, however, positive predictive values range from 26% to 37%.12,13
The Secretary of Health and Human Services Advisory Committee on Heritable Disorders in Newborns and Children created the Recommended Uniform Screening Panel14 to indicate the conditions for which screening should occur.15 The panel lists 34 core conditions, 25 of which are IEM15 (Table 215,16 ). The Recommended Uniform Screening Panel also lists 26 secondary conditions that can be detected on tandem mass spectrometry that are in the differential diagnosis of the core disorders.14 A number of criteria must be met for a disease to be added to the Recommended Uniform Screening Panel, including the ability of U.S. states to conduct the screening, evidence of a net benefit from screening, and availability of effective treatments. 17 Individual U.S. states control newborn screening; not all U.S. states screen for all core and secondary conditions.14,15
TABLE 2. Inborn Errors of Metabolism for Which Newborn Screening Is Recommended by the Secretary of Health and Human Services Advisory Committee on Heritable Disorders in Newborns and Children

| Condition listed by category | Clinical presentation |
|---|---|
| Amino acid disorder | |
| Classic phenylketonuria | Cognitive impairment, seizures, spasticity |
| Homocystinuria | Marfan-like appearance, lens dislocation, cognitive impairment, thromboembolism |
| Maple syrup urine disease | Progressive encephalopathy |
| Tyrosinemia, type I | Liver failure, septicemia, hypoglycemia, Fanconi syndrome (renal tubulopathy) |
| Fatty acid oxidation disorder | |
| Carnitine uptake defect/carnitine transport defect | Hypoketotic hypoglycemia, cardiomyopathy, liver disease |
| Long-chain L-3 hydroxyacyl-CoA dehydrogenase deficiency | Hypoketotic hypoglycemia, cardiomyopathy, liver disease, recurrent rhabdomyolysis |
| Medium-chain Acyl-CoA dehydrogenase deficiency | Reye-like syndrome, metabolic crisis after fasting with lethargy, nausea, vomiting, coma |
| Trifunctional protein deficiency | Hypoketotic hypoglycemia, cardiomyopathy, liver disease, recurrent rhabdomyolysis |
| Very long-chain Acyl-CoA dehydrogenase deficiency | Hypoketotic hypoglycemia, cardiomyopathy, liver disease, recurrent rhabdomyolysis |
| Organic acidemia | |
| 3-hydroxy-3-methylglutaric aciduria | Metabolic decompensation triggered by periods of fasting or infections |
| 3-methylcrotonyl-CoA carboxylase deficiency | Encephalopathy, ketoacidosis, hyperammonemia |
| ß-ketothiolase deficiency | Ketoacidotic attacks, sometimes leading to coma |
| Glutaric acidemia type I | Macrocephaly, encephalopathy |
| Holocarboxylase synthetase deficiency | Metabolic acidosis, neurologic defects, skin rash |
| Isovaleric acidemia | Encephalopathy, ketoacidosis, hyperammonemia |
| Methylmalonic acidemia (cobalamin disorders) | Encephalopathy, ketoacidosis, hyperammonemia |
| Methylmalonic acidemia (methylmalonyl-CoA mutase) | Encephalopathy, ketoacidosis, hyperammonemia |
| Propionic acidemia | Encephalopathy, ketoacidosis, hyperammonemia |
| Urea cycle disorders | |
| Argininosuccinic aciduria | Neurologic and liver abnormalities |
| Citrullinemia type I | Mild hyperammonemia, failure to thrive |
| Other | |
| Biotinidase deficiency | Metabolic acidosis, neurologic defects, skin rash |
| Classic galactosemia | Liver and renal failure after start of milk feeds in first week of life |
| Glycogen storage disease type II (Pompe disease) | Failure to thrive, cardiomyopathy, hypotonia |
| Mucopolysaccharidosis I | Hydrocephalus, cognitive impairment, coarse facies |
| X-linked adrenoleukodystrophy | Intellectual regression, leukodystrophy, adrenal insufficiency |
Many IEM in newborns are medical emergencies, and physicians should take immediate action if a positive newborn screen is received. The following actions should be taken: (1) contact the parents of the newborn to inform them of the situation; (2) assess the newborn's clinical condition; and (3) provide prompt referral to a metabolic specialist.16,18 The ACMG provides ACTion (ACT) sheet resources that describe the short-term actions a health professional should follow in communicating with the family and in determining the appropriate steps for an infant who has screened positive for an IEM disorder (see Medical Genetic Practice Resources at http://www.acmg.net).
Clinical Presentation
Various IEM can present throughout a patient's life span, from the prenatal period through adulthood.
PRENATAL
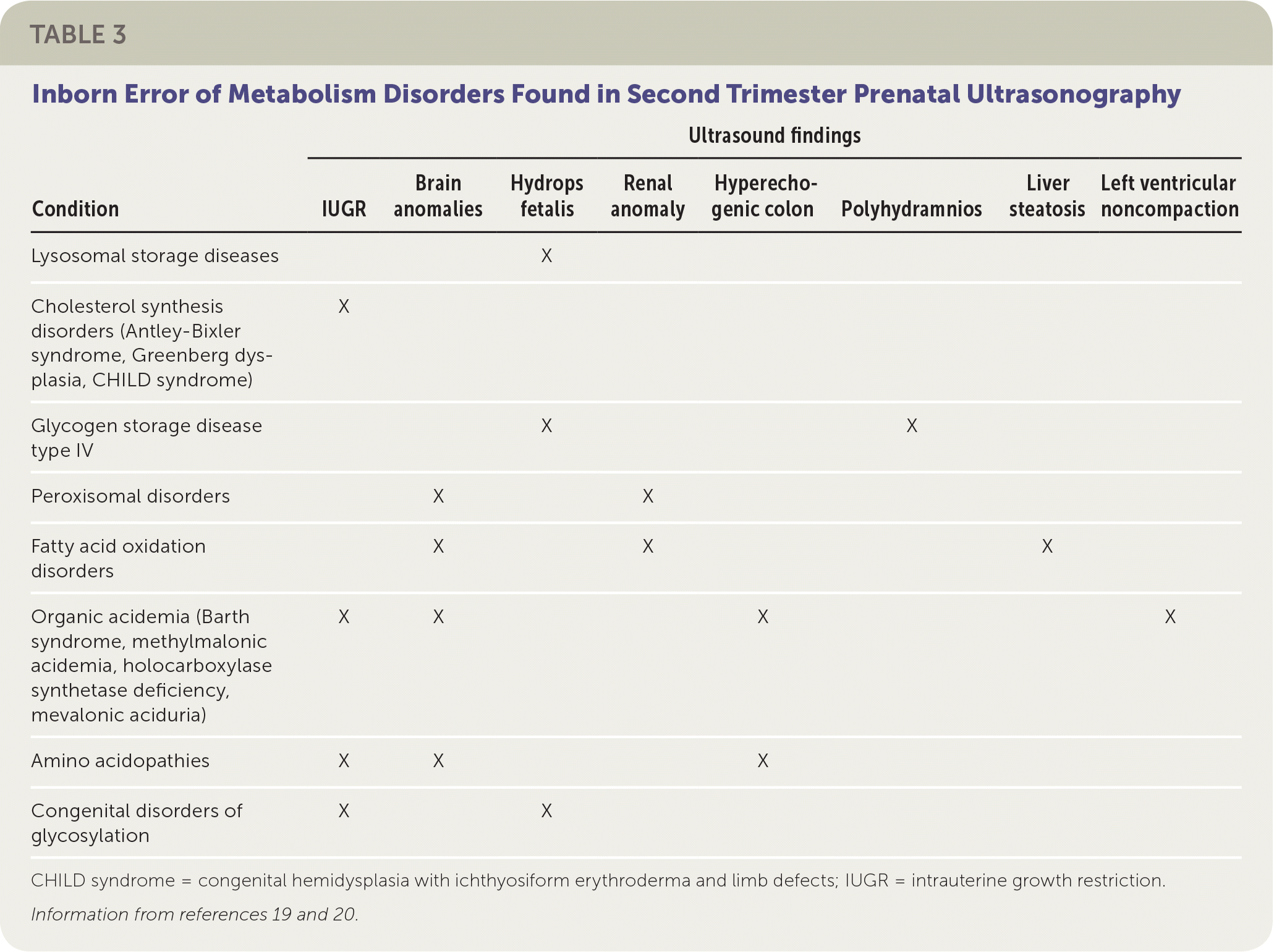
Some IEM present in pregnancy, with abnormalities detected on ultrasound. For example, hydrops fetalis (fluid accumulation in multiple body areas, such as ascites, pleural or pericardial effusions, or skin edema) may be seen on prenatal ultrasonography after 26 weeks' gestation in lysosomal storage diseases. Lysosomal storage diseases are a group of disorders that result from an enzyme deficiency that blocks the ability of lysosomes to digest large cellular molecules. The most common lysosomal storage diseases are mucopolysaccharidosis VII, Gaucher disease, and GM1 gangliosidosis.19 Other IEM presenting in utero on ultrasonograpy during the second trimester are shown in Table 3.19,20
TABLE 3. Inborn Error of Metabolism Disorders Found in Second Trimester Prenatal Ultrasonography
| Condition | Ultrasound findings | |||||||
|---|---|---|---|---|---|---|---|---|
| IUGR | Brain anomalies | Hydrops fetalis | Renal anomaly | Hyperechogenic colon | Polyhydramnios | Liver steatosis | Left ventricular noncompaction | |
| Lysosomal storage diseases | X | |||||||
| Cholesterol synthesis disorders (Antley-Bixler syndrome, Greenberg dysplasia, CHILD syndrome) | X | |||||||
| Glycogen storage disease type IV | X | X | ||||||
| Peroxisomal disorders | X | X | ||||||
| Fatty acid oxidation disorders | X | X | X | |||||
| Organic acidemia (Barth syndrome, methylmalonic acidemia, holocarboxylase synthetase deficiency, mevalonic aciduria) | X | X | X | X | ||||
| Amino acidopathies | X | X | X | |||||
| Congenital disorders of glycosylation | X | X | ||||||
CHILD syndrome = congenital hemidysplasia with ichthyosiform erythroderma and limb defects; IUGR = intrauterine growth restriction.
Maternal history, examination, and laboratory findings may be used in addition to fetal ultrasonography for indicators of IEM for two particular conditions. In ornithine transcarbamylase, the pregnant mother may be symptomatic with hyperammonemia, coma, and psychiatric symptoms.21 In long-chain hydroxyacyl dehydrogenase deficiency, the mother can present with an acute fatty liver; hyperemesis; and a syndrome with symptoms of hemolysis, elevated liver enzymes, and low platelet count.22
For all prenatal genetic diseases, whether detected with ultrasonography or suspected from clinical findings, a definitive diagnosis can be accomplished with chorionic villi sampling or amniocentesis.23
NEWBORN
Many IEM present in the first month of life. Often, the newborn will initially appear healthy because metabolites occurring in the IEM disorder have been cleared via placental circulation during the intrauterine period. Those metabolites accumulate only after birth; thus, a symptom-free period after birth is a vital component of the medical history.16
Symptoms of IEM in newborns are typically nonspecific, such as lethargy, poor feeding, vomiting, abnormal breathing, seizures, and/or hypotonia. Although these signs also signal infection (including sepsis), which is more common, IEM must be considered in the differential diagnosis. Additional findings that should raise concern about the possibility of an IEM disorder are metabolic acidosis, unexplained hypoglycemia, constitutional liver dysfunction, and encephalopathy.16
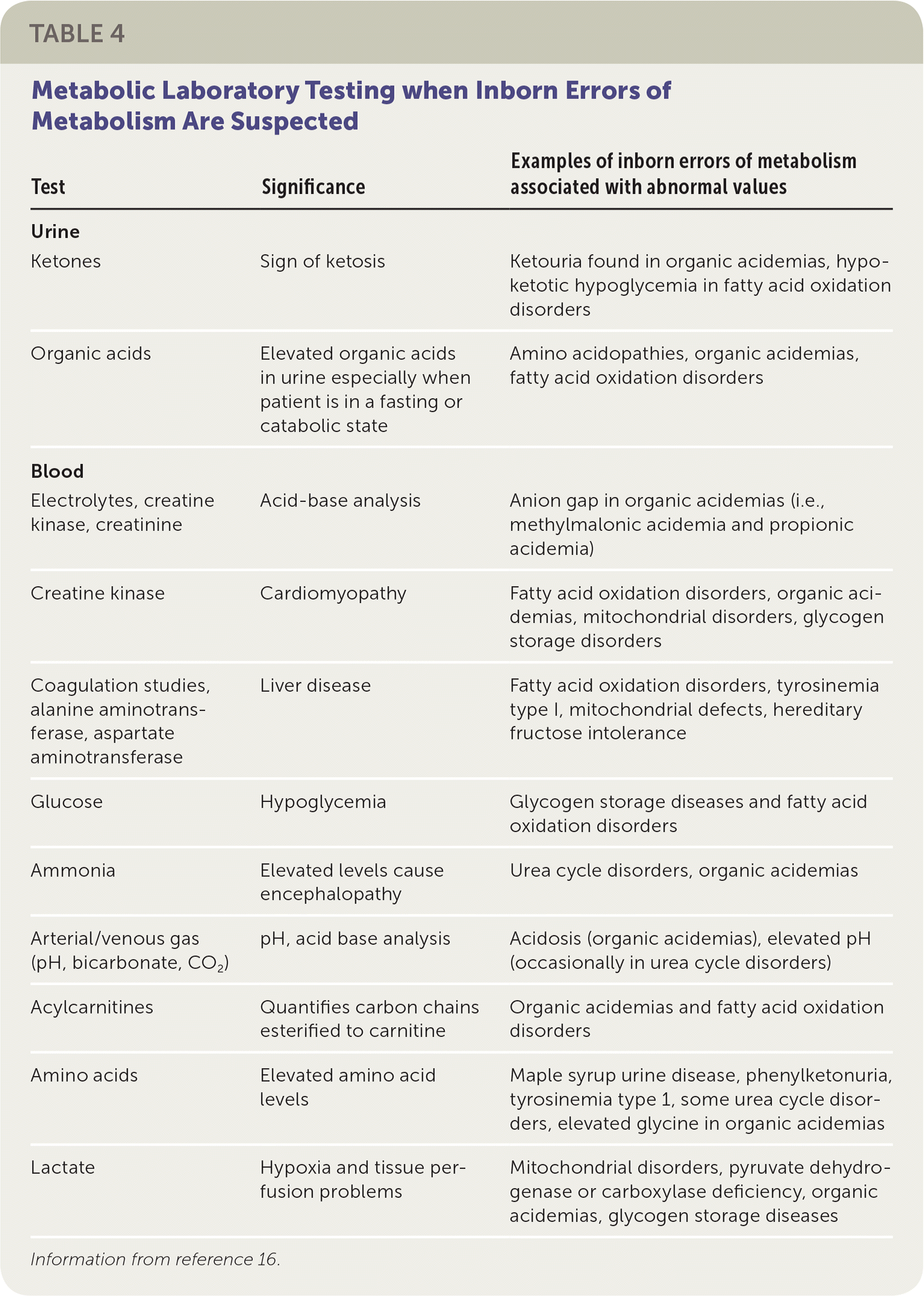
All newborns being evaluated for these presentations should have an ammonia level and urine ketone tests performed in addition to the evaluation for possible infection or sepsis. If acidosis is a concern (i.e., urine pH is low, respiratory rate is abnormal), a confirmation blood gas measurement should be performed, even if the pulse oximetry is normal. If any one of these tests is abnormal or if the newborn does not respond to antibiotics as expected,24,25 an IEM disorder should be considered, and a metabolic specialist should be consulted. If obtaining support from a metabolic specialist is delayed, plasma amino acids, acylcarnitine profile, and urine organic acids should be measured (Table 416 ).
TABLE 4. Metabolic Laboratory Testing when Inborn Errors of Metabolism Are Suspected
| Test | Significance | Examples of inborn errors of metabolism associated with abnormal values |
|---|---|---|
| Urine | ||
| Ketones | Sign of ketosis | Ketouria found in organic acidemias, hypoketotic hypoglycemia in fatty acid oxidation disorders |
| Organic acids | Elevated organic acids in urine especially when patient is in a fasting or catabolic state | Amino acidopathies, organic acidemias, fatty acid oxidation disorders |
| Blood | ||
| Electrolytes, creatine kinase, creatinine | Acid-base analysis | Anion gap in organic acidemias (i.e., methylmalonic acidemia and propionic acidemia) |
| Creatine kinase | Cardiomyopathy | Fatty acid oxidation disorders, organic acidemias, mitochondrial disorders, glycogen storage disorders |
| Coagulation studies, alanine aminotransferase, aspartate aminotransferase | Liver disease | Fatty acid oxidation disorders, tyrosinemia type I, mitochondrial defects, hereditary fructose intolerance |
| Glucose | Hypoglycemia | Glycogen storage diseases and fatty acid oxidation disorders |
| Ammonia | Elevated levels cause encephalopathy | Urea cycle disorders, organic acidemias |
| Arterial/venous gas (pH, bicarbonate, CO2) | pH, acid base analysis | Acidosis (organic acidemias), elevated pH (occasionally in urea cycle disorders) |
| Acylcarnitines | Quantifies carbon chains esterified to carnitine | Organic acidemias and fatty acid oxidation disorders |
| Amino acids | Elevated amino acidlevels | Maple syrup urine disease, phenylketonuria, tyrosinemia type 1, some urea cycle disorders, elevated glycine in organic acidemias |
| Lactate | Hypoxia and tissue perfusion problems | Mitochondrial disorders, pyruvate dehydrogenase or carboxylase deficiency, organic acidemias, glycogen storage diseases |
Information from reference 16.
Whereas many IEM present after a period during which the newborn appears healthy, some IEM may clinically present before the physician has received the IEM disorder screening test results. Additionally, a normal newborn metabolic screen should not deter physicians from performing a metabolic workup in a sick newborn because not all variations of metabolic diseases are covered in newborn screening tests.
CHILDREN
Most conditions that present in the newborn period can present during childhood in a similar or less severe manner and in some cases are linked with dysmorphic physical examination findings (Figure 1).26–28 Two of the most well-known examples are lysosomal storage disorders, Hunter syndrome and Hurler syndrome.29,30 The dysmorphic features may not present at birth, but they become more pronounced over time as the storage molecule accumulates.
FIGURE 1.
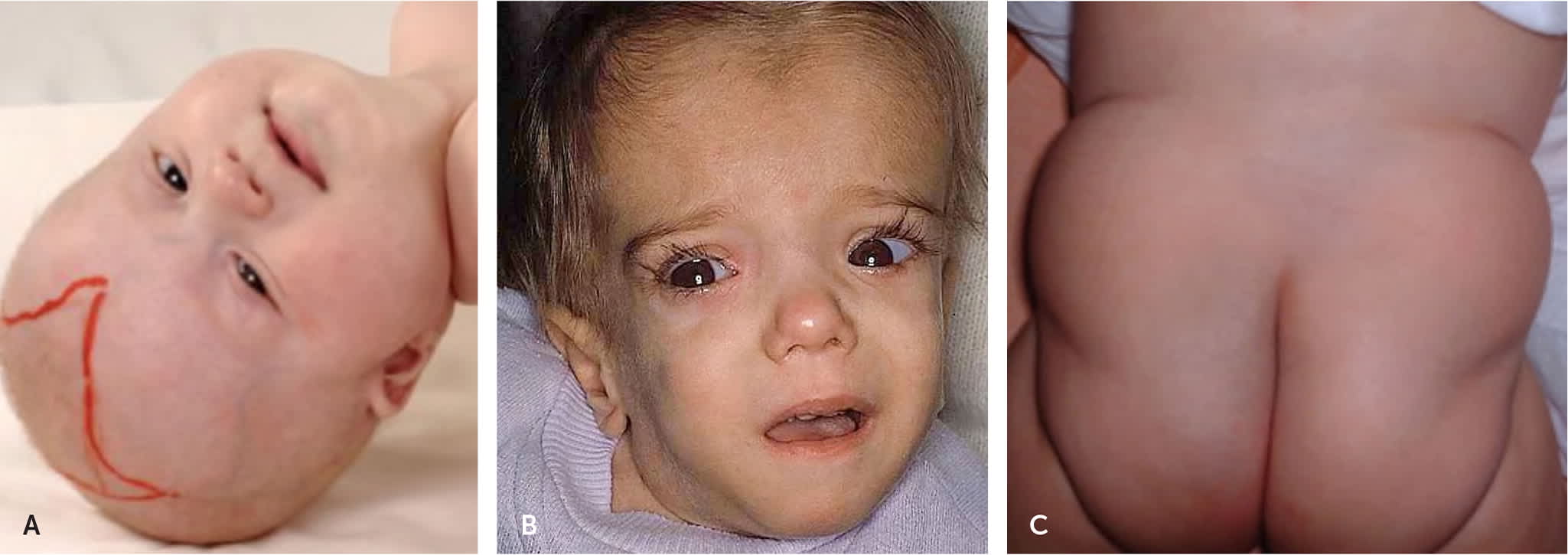
Recognizable dysmorphic features associated with inborn errors of metabolism disorders.
(A) Six-month-old girl with Zellweger syndrome,26 which is a leukodystrophy caused by a peroxisome biogenesis defect. Clinical features include epicanthal folds, high forehead, broad nasal bridge, hypoplastic supraorbital ridges, and enlarged anterior fontanel.
Reprinted with permission from Klouwer FC, Berendse K, Ferdinandusse S, et al. Zellweger spectrum disorders. Orphanet J Rare Dis. 2015;10:151.
(B) A 21-month-old child with mevalonic aciduria displaying typical dysmorphic features to include down-slanting eyes, blue sclerae, low set ears, and long face.27 Mevalonic aciduria presents with developmental delay, progressive ataxia, failure to thrive, visual defects, and periodic fevers.
Reprinted with permission from Haas D, Hoffmann GF. Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmoglobulinemia D syndrome. Orphanet J Rare Dis. 2006;1:13.
(C) Congenital disorders of glycosylation are a group of disorders with a disturbed ability to glycosylate certain tissue proteins and/or lipids, resulting in myriad presentations including seizures, ataxia, failure to thrive, liver disease, and brain anomalies. Lipodystrophy in a congenital disorder of glycosylation syndrome type 1a is shown.28
Reprinted with permission from Freeze HH, Eklund EA, Ng BG, Patterson MC. Neurology of inherited glycosylation disorders. Lancet Neurol. 2012;11(5):453–466.
ADOLESCENTS AND ADULTS
Primary care physicians occasionally encounter IEM in adolescents and adults (Table 531 ). Some children with IEM are living longer because of new treatments and a better understanding of how to avoid metabolic decompensation (e.g., avoiding high-protein diets in organic acidemia and urea cycle disorders). Also, some IEM have their onset in adults; for example, adult-onset lysosomal storage disorders. Patients with these IEM have residual enzyme activity that allows for slow accumulation of toxic molecules over time, and symptoms may not appear until adulthood. Some of these disorders (e.g., Gaucher disease) can be treated with enzyme replacement therapy.32
TABLE 5. Inborn Errors of Metabolism Presenting in Adolescents and Adults
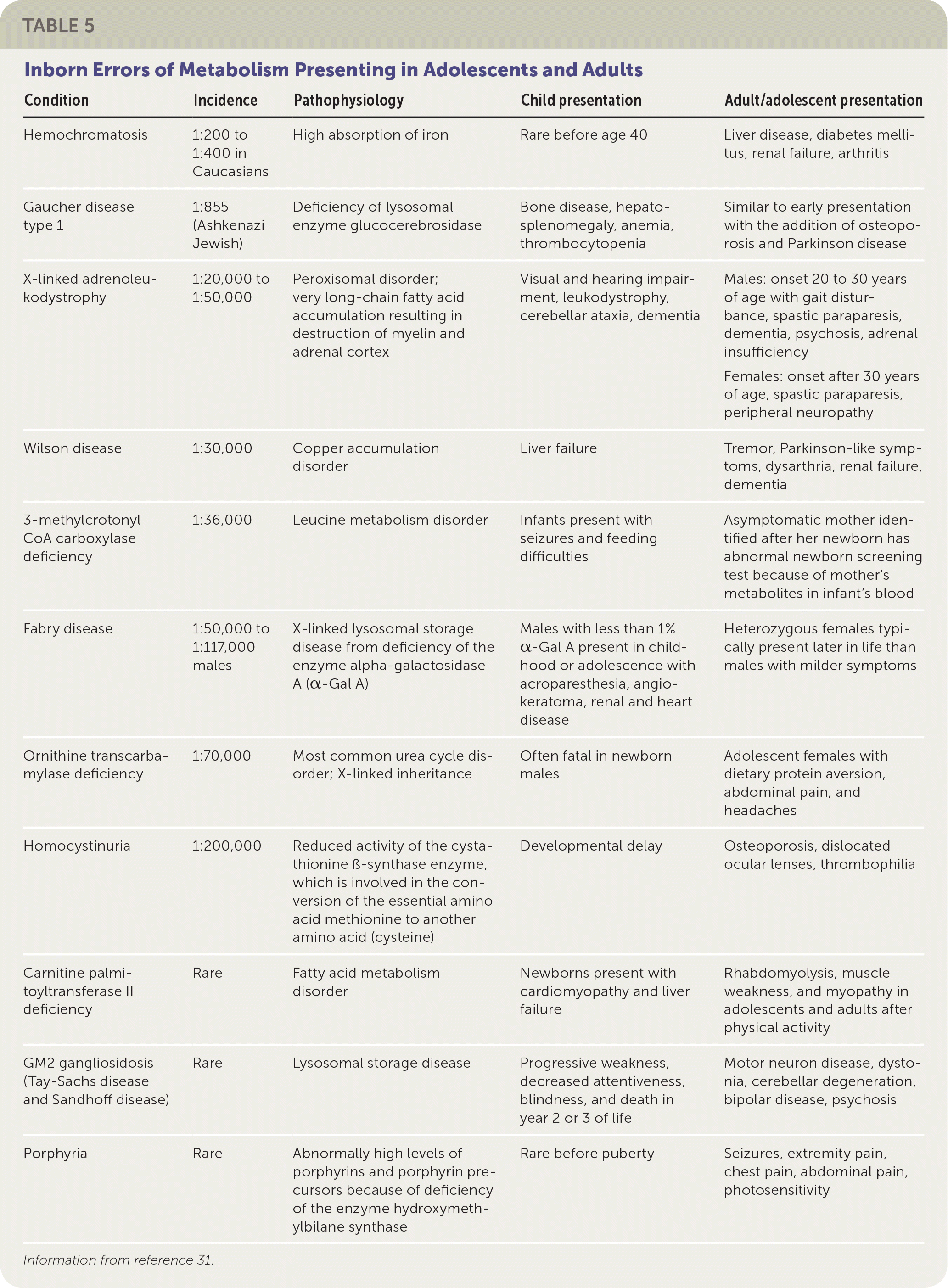
| Condition | Incidence | Pathophysiology | Child presentation | Adult/adolescent presentation |
|---|---|---|---|---|
| Hemochromatosis | 1:200 to 1:400 in Caucasians | High absorption of iron | Rare before age 40 | Liver disease, diabetes mellitus, renal failure, arthritis |
| Gaucher disease type 1 | 1:855 (Ashkenazi Jewish) | Deficiency of lysosomal enzyme glucocerebrosidase | Bone disease, hepatosplenomegaly, anemia, thrombocytopenia | Similar to early presentation with the addition of osteoporosis and Parkinson disease |
| X-linked adrenoleukodystrophy | 1:20,000 to 1:50,000 | Peroxisomal disorder; very long-chain fatty acid accumulation resulting in destruction of myelin and adrenal cortex | Visual and hearing impairment, leukodystrophy, cerebellar ataxia, dementia | Males: onset 20 to 30 years of age with gait disturbance, spastic paraparesis, dementia, psychosis, adrenal insufficiency Females: onset after 30 years of age, spastic paraparesis, peripheral neuropathy |
| Wilson disease | 1:30,000 | Copper accumulation disorder | Liver failure | Tremor, Parkinson-like symptoms, dysarthria, renal failure, dementia |
| 3-methylcrotonyl CoA carboxylase deficiency | 1:36,000 | Leucine metabolism disorder | Infants present with seizures and feeding difficulties | Asymptomatic mother identified after her newborn has abnormal newborn screening test because of mother's metabolites in infant's blood |
| Fabry disease | 1:50,000 to 1:117,000 males | X-linked lysosomal storage disease from deficiency of the enzyme alpha-galactosidase A (α-Gal A) | Males with less than 1% α-Gal A present in childhood or adolescence with acroparesthesia, angiokeratoma, renal and heart disease | Heterozygous females typically present later in life than males with milder symptoms |
| Ornithine transcarbamylase deficiency | 1:70,000 | Most common urea cycle disorder; X-linked inheritance | Often fatal in newborn males | Adolescent females with dietary protein aversion, abdominal pain, and headaches |
| Homocystinuria | 1:200,000 | Reduced activity of the cystathionine ß-synthase enzyme, which is involved in the conversion of the essential amino acid methionine to another amino acid (cysteine) | Developmental delay | Osteoporosis, dislocated ocular lenses, thrombophilia |
| Carnitine palmitoyltransferase II deficiency | Rare | Fatty acid metabolism disorder | Newborns present with cardiomyopathy and liver failure | Rhabdomyolysis, muscle weakness, and myopathy in adolescents and adults after physical activity |
| GM2 gangliosidosis (Tay-Sachs disease and Sandhoff disease) | Rare | Lysosomal storage disease | Progressive weakness, decreased attentiveness, blindness, and death in year 2 or 3 of life | Motor neuron disease, dystonia, cerebellar degeneration, bipolar disease, psychosis |
| Porphyria | Rare | Abnormally high levels of porphyrins and porphyrin pre-cursors because of deficiency of the enzyme hydroxymethylbilane synthase | Rare before puberty | Seizures, extremity pain, chest pain, abdominal pain, photosensitivity |
Information from reference 31.
Treatment
When physicians encounter a newborn or infant with acute decompensation that is attributable to an IEM disorder, the most important intervention is arranging urgent transfer to a center with a metabolic specialist.16,18 While awaiting transfer, the initial treatment is the same for all newborns and children with a suspected IEM disorder: stopping the buildup of toxic metabolites by discontinuing feedings and preventing catabolism by providing glucose at a high infusion rate (5 to 10 g per kg per hour).33,34 This is the equivalent of 10% dextrose in an electrolyte solution at 1.5 times maintenance rate or 5% dextrose at twice the maintenance rate. If this infusion leads to an elevation of serum lactate levels, it suggests the possibility of a mitochondrial disorder. If this occurs, the 10% dextrose fluid should be replaced with a lower glucose load, such as 5% dextrose.16
Although long-term treatment of IEM is beyond the scope of this review, note that chronic therapy is available for many IEM. The best known examples that respond to long-term management are the prototypical IEM disorder, PKU, and Gaucher disease. PKU can be treated with a restricted phenylalanine diet, and Gaucher disease can be treated with enzyme replacement. This demonstrates why accurate diagnosis is important.
This article updates a previous article on this topic by Raghuveer, et al.35
Data Sources: A PubMed search was completed in Clinical Queries using the key terms inherited metabolic disorders, genetic carrier screening, prenatal genetic testing, newborn screening, inborn errors of metabolism, and adults. The search included meta-analyses, clinical trials, and reviews. Also searched were the Agency for Healthcare Research and Quality evidence reports, the Cochrane database, the National Guideline Clear-inghouse, Bandolier, and Essential Evidence Plus. Search date: May 1, 2017.
The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the U.S. Department of Health and Human Services.
