Vitamin B12 (cobalamin) deficiency is a common cause of macrocytic anemia and has been implicated in a spectrum of neuropsychiatric disorders. The role of B12 deficiency in hyperhomocysteinemia and the promotion of atherosclerosis is only now being explored. Diagnosis of vitamin B12 deficiency is typically based on measurement of serum vitamin B12 levels; however, about 50 percent of patients with subclinical disease have normal B12 levels. A more sensitive method of screening for vitamin B12 deficiency is measurement of serum methylmalonic acid and homocysteine levels, which are increased early in vitamin B12 deficiency. Use of the Schilling test for detection of pernicious anemia has been supplanted for the most part by serologic testing for parietal cell and intrinsic factor antibodies. Contrary to prevailing medical practice, studies show that supplementation with oral vitamin B12 is a safe and effective treatment for the B12 deficiency state. Even when intrinsic factor is not present to aid in the absorption of vitamin B12 (pernicious anemia) or in other diseases that affect the usual absorption sites in the terminal ileum, oral therapy remains effective.
Vitamin B12 (cobalamin) plays an important role in DNA synthesis and neurologic function. Deficiency can lead to a wide spectrum of hematologic and neuropsychiatric disorders that can often be reversed by early diagnosis and prompt treatment.
The true prevalence of vitamin B12 deficiency in the general population is unknown. The incidence, however, appears to increase with age. In one study,1 15 percent of adults older than 65 years had laboratory evidence of vitamin B12 deficiency. The nearly ubiquitous use of gastric acid–blocking agents, which can lead to decreased vitamin B12 levels,2 may have an underappreciated role in the development of vitamin B12 deficiency. Taking the widespread use of these agents and the aging of the U.S. population into consideration, the actual prevalence of vitamin B12 deficiency may be even higher than statistics indicate. Despite these facts, the need for universal screening in older adults remains a matter of controversy.3,4
Clinical Manifestations
Vitamin B12 deficiency is associated with hematologic, neurologic, and psychiatric manifestations (Table 1). It is a common cause of macrocytic (megaloblastic) anemia and, in advanced cases, pancytopenia. Neurologic sequelae from vitamin B12 deficiency include paresthesias, peripheral neuropathy, and demyelination of the corticospinal tract and dorsal columns (subacute combined systems disease). Vitamin B12 deficiency also has been linked to psychiatric disorders, including impaired memory, irritability, depression, dementia and, rarely, psychosis.5,6
TABLE 1 Clinical Manifestations of Vitamin B12 Deficiency
| Hematologic |
| Megaloblastic anemia |
| Pancytopenia (leukopenia, thrombocytopenia) |
| Neurologic |
| Paresthesias |
| Peripheral neuropathy |
| Combined systems disease (demyelination of dorsal columns and corticospinal tract) |
| Psychiatric |
| Irritability, personality change |
| Mild memory impairment, dementia |
| Depression |
| Psychosis |
| Cardiovascular |
| Possible increased risk of myocardial infarction and stroke |
In addition to hematologic and neuropsychiatric manifestations, vitamin B12 deficiency may exert indirect cardiovascular effects. Similar to folic acid deficiency, vitamin B12 deficiency produces hyperhomocys-teinemia, which is an independent risk factor for atherosclerotic disease.7 Although the role of folic acid supplementation in reducing homocysteine levels as a method for preventing coronary artery disease and stroke continues to be a subject of great interest, there has been little emphasis on the potential role of vitamin B12 deficiency as a contributing factor in the development of cardiovascular disease. This possibility becomes especially important when considering vitamin replacement therapy. Folic acid supplementation may mask an occult vitamin B12 deficiency and further exacerbate or initiate neurologic disease. Therefore, clinicians should consider ruling out vitamin B12 deficiency before initiating folic acid therapy.8
Normal Absorption of Vitamin B12
In humans, only two enzymatic reactions are known to be dependent on vitamin B12. In the first reaction, methylmalonic acid is converted to succinyl-CoA using vitamin B12 as a cofactor (Figure 1). Vitamin B12 deficiency, therefore, can lead to increased levels of serum methylmalonic acid. In the second reaction, homocysteine is converted to methionine by using vitamin B12 and folic acid as cofactors. In this reaction, a deficiency of vitamin B12 or folic acid may lead to increased homocysteine levels.
FIGURE 1.
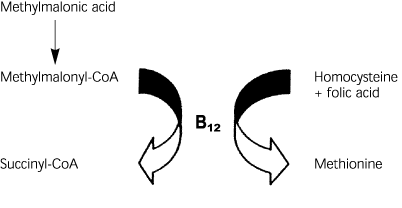
Vitamin B12 deficiency leads to a serum build-up of methylmalonic acid. Deficiency of vitamin B12 or folic acid can lead to increased homocysteine levels.
Information from Stabler SP. Screening the older population for cobalamin (vitamin B12) deficiency. J Am Geriatr Soc 1995;43:1291.
An understanding of the vitamin B12 absorption cycle helps illuminate the potential causes of deficiency. The acidic environment of the stomach facilitates the breakdown of vitamin B12 that is bound to food. Intrinsic factor, which is released by parietal cells in the stomach, binds to vitamin B12 in the duodenum. This vitamin B12–intrinsic factor complex subsequently aids in the absorption of vitamin B12 in the terminal ileum.
In addition to this method of absorption, evidence supports the existence of an alternate system that is independent of intrinsic factor or even an intact terminal ileum. Approximately 1 percent of a large oral dose of vitamin B12 is absorbed by this second mechanism.9 This pathway is important in relation to oral replacement. Once absorbed, vitamin B12 binds to transcobalamin II and is transported throughout the body. The interruption of one or any combination of these steps places a person at risk of developing deficiency (Figure 2).
FIGURE 2.
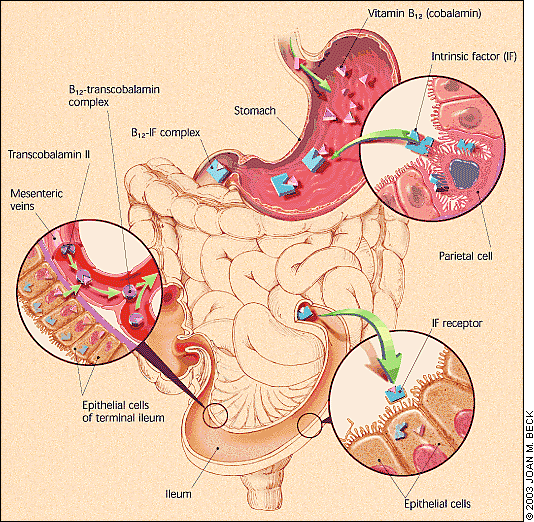
Vitamin B12 absorption and transport.
Diagnosis of Vitamin B12 Deficiency
The diagnosis of vitamin B12 deficiency has traditionally been based on low serum vitamin B12 levels, usually less than 200 pg per mL (150 pmol per L), along with clinical evidence of disease. However, studies indicate that older patients tend to present with neuropsychiatric disease in the absence of hematologic findings.5,6 Furthermore, measurements of metabolites such as methylmalonic acid and homocysteine have been shown to be more sensitive in the diagnosis of vitamin B12 deficiency than measurement of serum B12 levels alone.3,10–14
In a large study10 of 406 patients with known vitamin B12 deficiency, 98.4 percent had elevated serum methylmalonic acid levels, and 95.9 percent had elevated serum homocysteine levels (defined as three standard deviations above the mean). Only one patient out of 406 had normal levels of both metabolites, resulting in a sensitivity of 99.8 percent when methylmalonic acid and homocysteine levels are used for diagnosis. Interestingly, 28 percent of the patients in this study had normal hematocrit levels, and 17 percent had normal mean corpuscular volumes.
In another study13 of patients with known pernicious anemia who had not received maintenance vitamin B12 injections for months to years, the rise of methylmalonic acid and homocysteine levels was found to precede the decrease in serum vitamin B12 and the decline in hematocrit. This finding suggests that methylmalonic acid and homocysteine levels can be early markers for tissue vitamin B12 deficiency, even before hematologic manifestations occur.
Use of methylmalonic acid and homocysteine levels in the diagnosis of vitamin B12 deficiency has led to some surprising findings. If increased homocysteine or methylmalonic acid levels and a normalization of these metabolites in response to replacement therapy are used as diagnostic criteria for vitamin B12 deficiency, approximately 50 percent of these patients have serum vitamin B12 levels above 200 pg per mL.1 This observation suggests that use of a low serum vitamin B12 level as the sole means of diagnosis may miss up to one half of patients with actual tissue B12 deficiency. Other studies have shown similar findings, with the rate of missed diagnosis ranging from 10 to 26 percent when diagnosis is based on low serum vitamin B12 levels alone.3
There are, however, a few caveats to keep in mind. Looking at the reactions that use vitamin B12 (Figure 1),3 an elevated methylmalonic acid level is clearly more specific for vitamin B12 deficiency than an elevated homocysteine level. Vitamin B12 or folic acid deficiency can cause the homocysteine level to rise, so folic acid levels also should be checked in patients with isolated hyperhomocysteinemia.
In addition, folic acid deficiency can cause falsely low serum vitamin B12 levels. One study14 revealed that approximately one third of patients with folic acid deficiency had low serum vitamin B12 levels—less than 100 pg per mL (74 pmol per L) in some patients. Also, methylmalonic acid levels can be elevated in patients with renal disease (the result of decreased urinary excretion); thus, elevated levels must be interpreted with caution.10
FIGURE 3. Suspected Vitamin B12 Deficiency
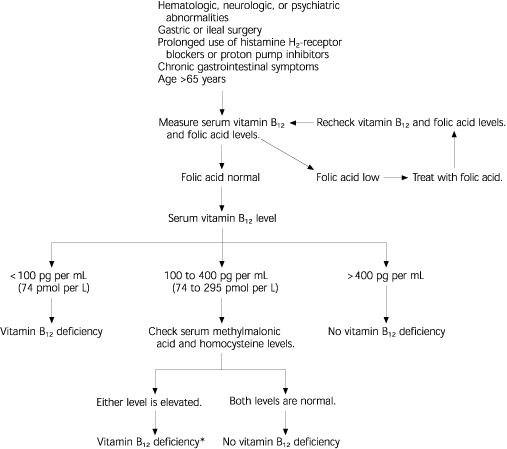
Suggested approach to the patient with suspected vitamin B12 deficiency.
*-If levels of metabolites normalize with vitamin B12 supplementation.
Information from Stabler SP. Screening the older population for cobalamin (vitamin B12) deficiency. J Am Geriatr Soc1995:43:1295, and Snow CF. Laboratory diagnosis of vitamin B12 and folate deficiency. Arch Intern Med 1999;159:1297.
Causes of Vitamin B12 Deficiency States
Once vitamin B12 deficiency is confirmed, a search for the etiology should be initiated. Causes of vitamin B12 deficiency can be divided into three classes: nutritional deficiency, malabsorption syndromes, and other gastrointestinal causes (Table 2).14
NUTRITIONAL DEFICIENCY
Dietary sources of vitamin B12 are primarily meats and dairy products. In a typical Western diet, a person obtains approximately 5 to 15 mcg of vitamin B12 daily, much more than the recommended daily allowance of 2 mcg. Normally, humans maintain a large vitamin B12 reserve, which can last two to five years even in the presence of severe malab-sorption.14 Nevertheless, nutritional deficiency can occur in specific populations. Elderly patients with “tea and toast” diets and chronic alcoholics are at especially high risk. The dietary limitations of strict vegans make them another, less common at-risk population.
MALABSORPTION SYNDROMES
The classic disorder of malabsorption is pernicious anemia, an autoimmune disease that affects the gastric parietal cells. Destruction of these cells curtails the production of intrinsic factor and subsequently limits vitamin B12 absorption. Laboratory evidence of parietal cell antibodies is approximately 85 to 90 percent sensitive for the diagnosis of pernicious anemia. However, the presence of parietal cell antibodies is nonspecific and occurs in other autoimmune states. Intrinsic factor antibody is only 50 percent sensitive, but it is far more specific for the diagnosis of pernicious anemia.
A Schilling test, which distinguishes intrinsic factor-related malabsorption, can be used to diagnose pernicious anemia (Table 3).14 Specifically, Schilling test results were once used to determine whether a patient required parenteral or oral vitamin B12 supplementation. This distinction is now unnecessary, because evidence points to a B12 absorption pathway independent of intrinsic factor, and studies have proved that oral replacement is equal in efficacy to intramuscular therapy.9 Regardless of the test result, successful treatment can still be achieved with oral replacement therapy.
Thus, the utility of the Schilling test has been brought into question.3 The Schilling test also has fallen out of favor because it is complicated to perform, the radiolabeled vitamin B12 is difficult to obtain, and interpretation of test results can be problematic in patients with renal insufficiency.
The phenomenon of food-bound malab-sorption occurs when vitamin B12 bound to protein in foods cannot be cleaved and released. Any process that interferes with gastric acid production can lead to this impairment. Atrophic gastritis, with resulting hypochlorhydria, is a major cause, especially in the elderly.3 Subtotal gastrectomy, once common before the availability of effective medical therapy for peptic ulcer disease, also can lead to vitamin B12 deficiency by this mechanism.
As mentioned previously, the widespread and prolonged use of histamine H2-receptor blockers and proton pump inhibitors for ulcer disease also may cause impaired breakdown of vitamin B12 from food, causing malabsorption and eventual depletion of B12 stores. Recent studies have confirmed that long-term use of omeprazole can lead to lower serum vitamin B12 levels.15,16 While more studies are needed to identify the incidence and prevalence of vitamin B12 deficiency in this subset of patients, screening for subclinical B12 deficiency should be a consideration in patients who have received long-term acid-suppression therapy.2
OTHER CAUSES
Other etiologies of vitamin B12 deficiency, although less common, deserve mention. Patients with evidence of vitamin B12 deficiency and chronic gastrointestinal symptoms such as dyspepsia, recurrent peptic ulcer disease, or diarrhea may warrant evaluation for such entities as Whipple's disease (a rare bacterial infection that impairs absorption), Zollinger-Ellison syndrome (gastrinoma causing peptic ulcer and diarrhea), or Crohn's disease. Patients with a history of intestinal surgery, strictures, or blind loops may have bacterial overgrowth that can compete for dietary vitamin B12 in the small bowel, as can infestation with tapeworms or other intestinal parasites. Congenital transport-protein deficiencies, including transcobalamin II deficiency, are another rare cause of vitamin B12 deficiency.
Oral vs. Parenteral Therapy
Because most clinicians are generally unaware that oral vitamin B12 therapy is effective,17 the traditional treatment for B12 deficiency has been intramuscular injections. However, since as early as 1968, oral vitamin B12 has been shown to have an efficacy equal to that of injections in the treatment of pernicious anemia and other B12 deficiency states.9,17–19 Although the majority of dietary vitamin B12 is absorbed in the terminal ileum through a complex with intrinsic factor, evidence for the previously mentioned alternate transport system is mounting.
In one study,18 38 patients with vitamin B12 deficiency were randomized to receive oral or parenteral therapy. Patients in the parenteral therapy group received 1,000 mcg of vitamin B12 intramuscularly on days 1, 3, 7, 10, 14, 21, 30, 60, and 90, while those in the oral treatment group received 2,000 mcg daily for 120 days. At the end of 120 days, patients who received oral therapy had significantly higher serum vitamin B12 levels and lower methyl-malonic acid levels than those in the parenteral therapy group. The actual transport mechanism used in this pathway remains unproved, but vitamin B12 is thought to be absorbed “en masse” in high doses. Surprisingly, one study20 showed that even in patients who had undergone gastrectomy, vitamin B12 deficiency could be easily reversed with oral supplementation.
Intramuscular injections, although safe and inexpensive, have several drawbacks. Injections are painful, medical personnel giving the injections are placed at risk of needlestick injuries, and administration of intramuscular injections often adds to the cost of therapy. Treatment schedules for intramuscular administration vary widely but usually consist of initial loading doses followed by monthly maintenance injections. One regimen consists of daily injections of 1,000 mcg for one to two weeks, then a maintenance dose of 1,000 mcg every one to three months.
Although the daily requirement of vitamin B12 is approximately 2 mcg, the initial oral replacement dosage consists of a single daily dose of 1,000 to 2,000 mcg (Table 4). This high dose is required because of the variable absorption of oral vitamin B12 in doses of 500 mcg or less.19 This regimen has been shown to be safe, cost-effective, and well tolerated by patients.19
TABLE 4 Schedule for Vitamin B12 Therapy
| Route of administration | Initial dosage | Maintenance dosage |
|---|---|---|
| Oral | 1,000 to 2,000 mcg per day for one to two weeks | 1,000 mcg per day for life |
| Intramuscular | 100 to 1,000 mcg every day or every other day for one to two weeks | 100 to 1,000 mcg every one to three months |
Follow-Up
After the diagnosis of vitamin B12 deficiency has been made and a treatment plan has been initiated, follow-up is important to determine the patient's response to therapy. If vitamin B12 deficiency is associated with severe anemia, correction of the deficiency state should lead to a marked reticulocytosis in one to two weeks. In mild vitamin B12 deficiency, we recommend repeat measurements of serum vitamin B12, homocysteine, and methylmalonic acid levels two to three months after initiating treatment.
