Hematologic emergencies are bleeding or clotting disorders that are hereditary or acquired and must be treated emergently to avoid significant morbidity or mortality. Patients experiencing a hematologic emergency may present with spontaneous bleeding, jaundice, petechiae, or purpura. Initial diagnostic testing should include a complete blood count. Patients who have bleeding associated with a hereditary disorder should receive clotting factor replacement before diagnostic testing. Acute chest syndrome is an uncommon but serious complication of sickle cell disease. Hemolysis caused by autoimmune disorders or iatrogenic errors from blood product transfusions has a distinct clinical presentation and requires immediate action. Severe thrombocytopenia presenting as immune or thrombotic thrombocytopenic purpura should be differentiated and treated appropriately. Disseminated intravascular coagulation and trauma coagulopathy are sometimes confused with each other, but both can cause serious injury and require unique treatments. Primary care physicians should promptly recognize patients who require emergent referral to a hematologic specialist.
Hematologic emergencies are bleeding or clotting disorders that must be treated emergently to avoid significant morbidity or mortality. Hematologic emergencies can be hereditary or acquired and subdivided into bleeding, clotting, or combined disorders. This article covers selected hematologic conditions that require urgent evaluation. Bleeding emergencies present with clinical symptoms and often significant laboratory findings such as thrombocytopenia (platelets less than 10 × 103 per μL), elevated international normalized ratio [INR], or anemia with evidence of hemolysis (Table 11–4). Blood product transfusions, including platelets, packed red blood cells (RBCs), cryoprecipitate, and factors VIII, IX, or VII, are critical in treating bleeding emergencies (Table 2).5–7 Corticosteroids, monoclonal antibodies, intravenous immunoglobulin (Ig), and exchange transfusions may be needed for acquired hematologic emergencies, in addition to blood products. During a potential hematologic emergency, a hematologist referral should be considered when active bleeding requires the administration of hemostatic products, abnormal findings on initial blood level testing cannot be easily explained, or an acute hematologic malignancy is suspected.
SORT: KEY RECOMMENDATIONS FOR PRACTICE
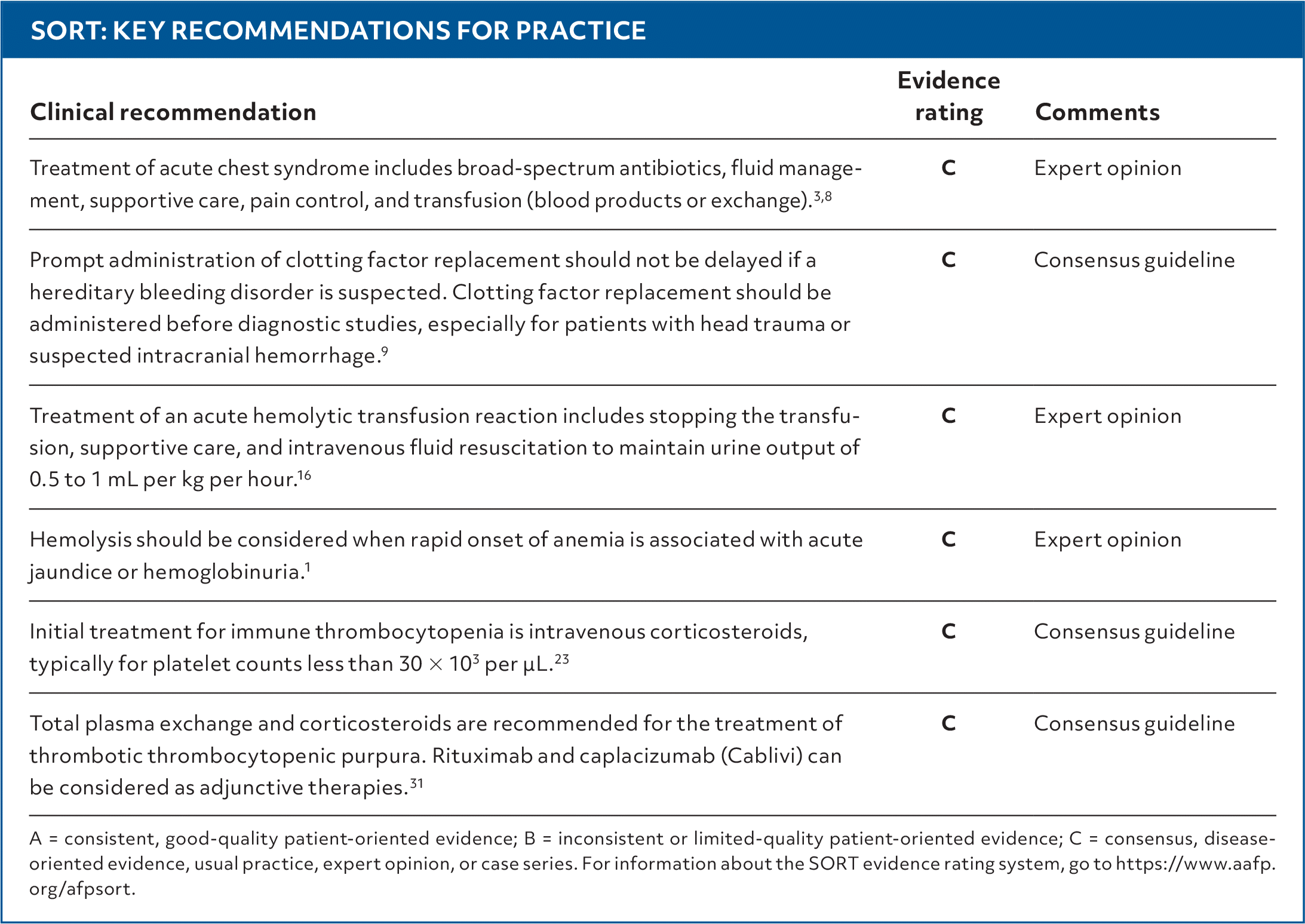
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.
TABLE 1. Clinical History and Laboratory Findings in Specific Hematologic Emergencies
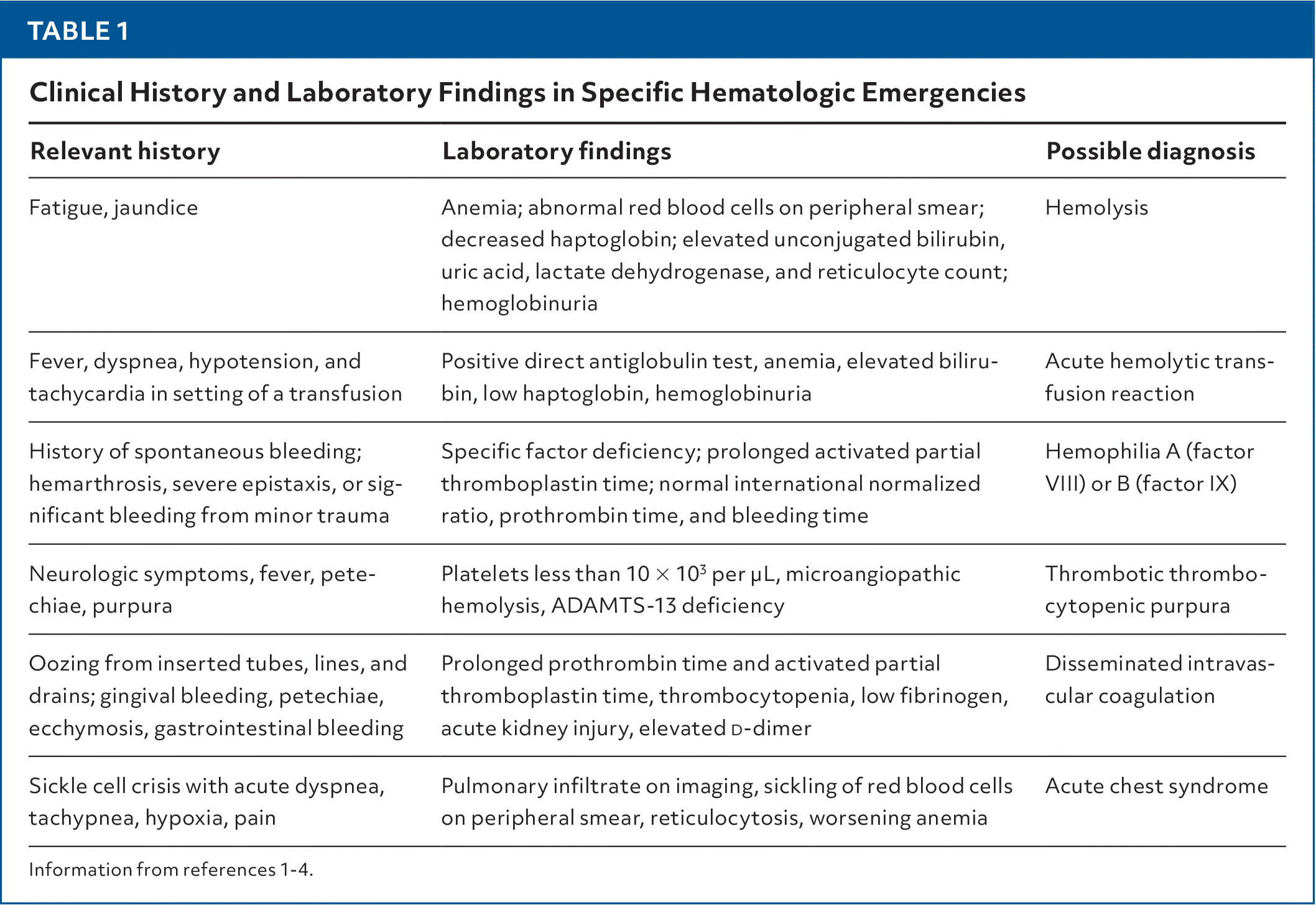
| Relevant history | Laboratory findings | Possible diagnosis |
|---|---|---|
| Fatigue, jaundice | Anemia; abnormal red blood cells on peripheral smear; decreased haptoglobin; elevated unconjugated bilirubin, uric acid, lactate dehydrogenase, and reticulocyte count; hemoglobinuria | Hemolysis |
| Fever, dyspnea, hypotension, and tachycardia in setting of a transfusion | Positive direct antiglobulin test, anemia, elevated bilirubin, low haptoglobin, hemoglobinuria | Acute hemolytic transfusion reaction |
| History of spontaneous bleeding; hemarthrosis, severe epistaxis, or significant bleeding from minor trauma | Specific factor deficiency; prolonged activated partial thromboplastin time; normal international normalized ratio, prothrombin time, and bleeding time | Hemophilia A (factor VIII) or B (factor IX) |
| Neurologic symptoms, fever, petechiae, purpura | Platelets less than 10 × 103 per μL, microangiopathic hemolysis, ADAMTS-13 deficiency | Thrombotic thrombocytopenic purpura |
| Oozing from inserted tubes, lines, and drains; gingival bleeding, petechiae, ecchymosis, gastrointestinal bleeding | Prolonged prothrombin time and activated partial thromboplastin time, thrombocytopenia, low fibrinogen, acute kidney injury, elevated | Disseminated intravascular coagulation |
| Sickle cell crisis with acute dyspnea, tachypnea, hypoxia, pain | Pulmonary infiltrate on imaging, sickling of red blood cells on peripheral smear, reticulocytosis, worsening anemia | Acute chest syndrome |
TABLE 2. Blood Products for Transfusion and Factor Replacement
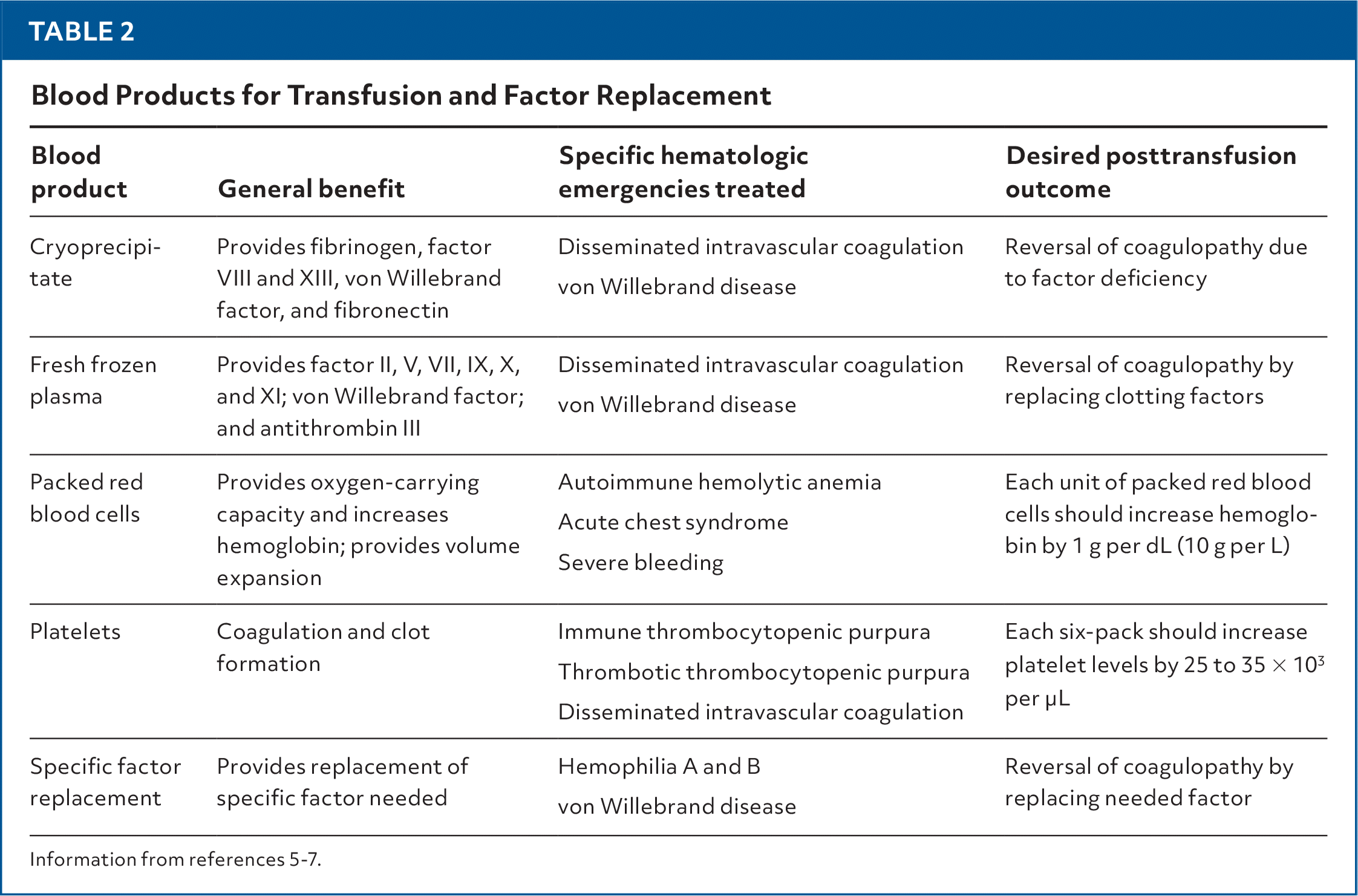
| Blood product | General benefit | Specific hematologic emergencies treated | Desired posttransfusion outcome |
|---|---|---|---|
| Cryoprecipitate | Provides fibrinogen, factor VIII and XIII, von Willebrand factor, and fibronectin | Disseminated intravascular coagulation von Willebrand disease | Reversal of coagulopathy due to factor deficiency |
| Fresh frozen plasma | Provides factor II, V, VII, IX, X, and XI; von Willebrand factor; and antithrombin III | Disseminated intravascular coagulation von Willebrand disease | Reversal of coagulopathy by replacing clotting factors |
| Packed red blood cells | Provides oxygen-carrying capacity and increases hemoglobin; provides volume expansion | Autoimmune hemolytic anemia Acute chest syndrome Severe bleeding | Each unit of packed red blood cells should increase hemoglobin by 1 g per dL (10 g per L) |
| Platelets | Coagulation and clot formation | Immune thrombocytopenic purpura Thrombotic thrombocytopenic purpura Disseminated intravascular coagulation | Each six-pack should increase platelet levels by 25 to 35 × 103 per μL |
| Specific factor replacement | Provides replacement of specific factor needed | Hemophilia A and B von Willebrand disease | Reversal of coagulopathy by replacing needed factor |
HEREDITARY DISORDERS
Acute Chest Syndrome in Sickle Cell Disease
Acute chest syndrome is an uncommon and emergent complication of an acute sickle cell crisis that requires immediate action and has a mortality of 9% in adults.8 The diagnosis is made when a patient with sickle cell disease has a new pulmonary infiltrate on chest radiography or computed tomography with clinical findings of fever, chest pain, respiratory distress, hypoxemia, cough, or tachypnea. Differentiating acute chest syndrome from pneumonia, pulmonary embolus, or acute coronary syndrome is challenging on initial presentation.8 Acute chest syndrome characteristically follows an acute sickle cell pain crisis by 24 to 72 hours.
Known etiologies include infection, fat emboli, and direct vaso-occlusive disease of the lungs. Fat emboli are definitively diagnosed when greater than 5% of lipid-laden macrophages are visible on bronchoscopy or sputum samples. However, bronchoscopy is not usually performed in this population due to an increased risk of mechanical ventilation requirements.3,8
Treatment for acute chest syndrome includes broad-spectrum antibiotics, fluid management, supportive care, pain control, and transfusion (standard blood products or an exchange transfusion).3,8 Exchange transfusion is reserved for more severe acute chest syndrome with multilobar pulmonary involvement, rapidly developing respiratory distress, or hemoglobin levels greater than 9 g per dL (90 g per L). Patients with a platelet count less than 200 × 103 per μL, multilobar involvement, and a history of coronary artery disease are at higher risk of requiring mechanical ventilation and should be closely observed. Hydroxyurea is an important prophylactic medication that has been shown to reduce acute chest syndrome incidence by 50% and overall sickle cell disease mortality by 40%.8 Hydroxyurea is teratogenic and contraindicated in those who are pregnant or breastfeeding.8
Bleeding Disorders
Severe hereditary bleeding disorders occur when clotting factors are absent or extremely deficient (baseline factor levels less than 1%).9 Severe bleeding can occur spontaneously or secondary to physical trauma. The three most common congenital bleeding disorders are hemophilia A (factor VIII deficiency), hemophilia B (factor IX deficiency), and von Willebrand disease. Although von Willebrand disease worsens the ability of platelets to form a clot, many patients with von Willebrand disease have factor VIII: C deficiencies.
Severe hereditary factor deficiencies are typically diagnosed at birth or shortly after with findings of intracranial hemorrhage, cephalohematomas, or significant bleeding at the circumcision site.10 Patients with hereditary bleeding disorders can also exhibit significant bruising, gingival bleeding, severe menorrhagia, and hematomas from minor trauma. The workup for hereditary bleeding disorders includes obtaining prothrombin time (PT), INR, activated partial thromboplastin time (PTT), platelet count, and bleeding time and completing bleeding assessment tools. However, bleeding time is not routinely used because of a lack of adequate standardization.11 Activated PTT is prolonged when factors VIII or IX are deficient; therefore, patients with hemophilia A or B have prolonged activated PTT but normal PT, INR, and bleeding time. Von Willebrand disease presents as an increase in bleeding time and a deficiency in von Willebrand factor, but patients have a normal PT and INR. Activated PTT is also typically normal but may be mildly elevated if there are low levels of factor VIII.4,10
The National Bleeding Disorders Foundation provides specific guidelines on emergent assessment and treatment for patients with suspected bleeding disorders.9 Clotting factor replacement based on the percentage of factor desired should be initiated when bleeding is suspected, even before diagnostic tests (e.g., clotting factor levels, head computed tomography in suspected intracranial hemorrhage, radiography of joints with potential hemarthrosis).
Table 3 summarizes indications for factor replacement or transfusion.9 When bleeding is considered severe (needing 80% to 100% replacement), patients with hemophilia A should receive replacement factor VIII at 50 units per kg, and patients with hemophilia B should receive factor IX at 100 to 140 units per kg. In less severe bleeding, lower doses may be appropriate.
TABLE 3. Indications for Factor Replacement or Transfusion in a Patient With Known Hereditary Bleeding Disorder

| Acute fractures, dislocations, and sprains |
| Gastrointestinal bleeding leading to moderate or severe anemia |
| Heavy menstrual bleeding leading to moderate or severe anemia or volume instability |
| Heavy or persistent bleeding from any site |
| History of an accident or trauma that might result in internal bleeding |
| Localized severe pain or swelling |
| New or unusual headache, particularly following trauma |
| Open wounds that require surgical closure, wound adhesive, or wound closure strips (e.g., Steri-Strips) |
| Recent invasive procedure or surgery |
| Significant injury to the head, neck, mouth, or eyes or evidence of bleeding in these areas |
| Suspected significant bleeding into a joint or muscle |
Adapted with permission from the National Bleeding Disorders Foundation. MASAC document 257 – guidelines for emergency department management of individuals with hemophilia and other bleeding disorders. December 5, 2019. Accessed February 25, 2023. https://www.hemophilia.org/healthcare-professionals/guidelines-on-care/masac-documents/masac-document-257-guidelines-for-emergency-department-management-of-individuals-with-hemophilia-and-other-bleeding-disorders.
Cryoprecipitate and fresh frozen plasma are not recommended for patients with hemophilia A or B. Patients with von Willebrand disease may be treated with synthetic desmopressin for minor bleeding, but bleeding emergencies in these patients require transfusion of factor VIII and von Willebrand factor concentrate.10,12 Cryoprecipitate and fresh frozen plasma have adequate factor VIII and von Willebrand factor, but due to the small amount of each per transfused unit, patients require multiple units to reverse von Willebrand disease bleeding. Patients with known hereditary bleeding disorders often wear medical alert bracelets and travel with their own factor replacement and treatment plan from their hemophilia treatment center. The National Bleeding Disorders Foundation recommends using factor products provided by patients when possible, instead of hospital-provided products, to maximize treatment speed and safety.9
ACQUIRED DISORDERS
Acquired Hemophilia A
Although hereditary hemophilia is more common in all people, acquired hemophilia A presents more commonly in people older than 60 years or postpartum and has a 20% lifetime mortality risk. It is caused by neutralizing autoantibodies to factor VIII.13 These antibodies are associated with infection, malignancy, pregnancy, or other autoimmune disorders. The clinical presentation of acquired hemophilia is similar to the hereditary type, and hemorrhage should be treated aggressively with bypassing agents, including recombinant VIIa, activated prothrombin complex concentrates, or recombinant porcine factor VIII.13
After the initial hemorrhage is controlled, high-dose corticosteroids (e.g., 1 mg per kg of prednisone with rituximab or cyclophosphamide) are used for autoantibody inhibition. Clinical suspicion for acquired hemophilia A should be high in patients with risk factors and an elevated activated PTT, especially when the residual factor VIII levels are higher than expected, with an associated hemorrhage.13
Acute Hemolytic Transfusion Reaction
Reactions to blood product infusions are common, but most are not severe. An acute hemolytic transfusion reaction is a hematologic emergency that may follow the transfusion of ABO or non-ABO antigen-incompatible RBCs or plasma.14 The classic triad of symptoms is fever, renal pain, and hemoglobinuria; other symptoms may include severe intravenous site pain, nausea, vomiting, headache, tachypnea, tachycardia, pallor, edema, and dyspnea.15,16 In patients who are sedated, evidence of shock and hemoglobinuria may be the only signs of acute hemolytic transfusion reaction.15
Patients with an acute hemolytic transfusion reaction begin bleeding due to the development of disseminated intravascular coagulation while also experiencing renal failure secondary to shock and excessive myoglobin deposits. A laboratory evaluation should include a direct antiglobulin test, lactate dehydrogenase, haptoglobin, bilirubin, complete blood count, and a basic metabolic panel, with particular attention to potential hyperkalemia.15
Treatment of acute hemolytic transfusion reaction includes stopping the transfusion; providing supportive care, including vasopressors to support blood pressure; and using intravenous fluid resuscitation to maintain a urine output of 0.5 to 1 mL per kg per hour.16 Case studies of severe hemolytic reactions have shown varying success with monoclonal antibody treatment or plasma exchange transfusions.16
Autoimmune Hemolytic Anemia
Autoimmune hemolytic anemia (AIHA) occurs when autoantibodies attack RBCs, leading to lysis and inadequate functional capacity. AIHA can manifest as a severe, symptomatic anemia unresponsive to a packed RBC transfusion. The diagnosis is confirmed in patients with a positive direct antiglobulin test result and severe anemia without other defined causes of a positive direct antiglobulin test result. Elevated reticulocyte count, lactate dehydrogenase, or bilirubin or decreased haptoglobin is common.17
AIHA can be classified as warm or cold, depending on the temperature stability of the autoantibodies.17 Cold agglutinins are specific to IgM, and warm agglutinins are specific to IgG antibodies. Warm AIHA is more common and associated with extravascular hemolysis, whereas cold AIHA is associated with intravascular hemolysis and higher lactate dehydrogenase levels.18 Warm AIHA has a more fulminant course and is considered a more severe form of hemolysis, with a mortality rate of up to 22%.18
Patients present with fatigue on exertion, dark urine, and acute jaundice.1 Treatment consists of corticosteroids (e.g., prednisone, 100 to 200 mg per day) and monoclonal antibodies (e.g., rituximab), with a 70% to 80% response rate.19,20 Emergent packed RBC transfusion is necessary for patients with hemoglobin less than 7 g per dL (70 g per L) or reticulocytopenia, even though many RBCs can undergo hemolysis following transfusion.19 It is also important to warm packed RBCs to a normal body temperature before administering them to a patient with cold AIHA, because this can lead to tolerance of transfusions without additional hemolysis in more than 50% of patients.21,22 Working in conjunction with the blood product laboratory is essential because immunomodulation treatment is critical to slowing hemolysis and achieving an effective transfusion in severe AIHA.
For resistant AIHA, splenectomy is an effective and definitive treatment.19 Other less proven medical therapies for AIHA include intravenous IgG, cyclophosphamide, and hematopoietic stem cell transplant. See the 2018 American Family Physician article for more on hemolytic anemias.1
Immune Thrombocytopenic Purpura
Immune thrombocytopenic purpura is the most common cause of isolated thrombocytopenia and results from autoantibodies attacking platelets (Table 42,23–26). Immune thrombocytopenic purpura typically presents without symptoms of systemic illness and with an otherwise normal complete blood count. Although normal platelet counts range from 150 to 450 × 103 per μL, severe thrombocytopenia is commonly defined as platelet counts less than 20 × 103 per μL. Patients with platelet counts less than 10 × 103 per μL are likely to have spontaneous bleeding, visible petechiae, and purpura. These patients are at high risk of life-threatening bleeding, and platelet transfusion is indicated with active bleeding.
TABLE 4. Emergent Thrombocytopenic Disorders
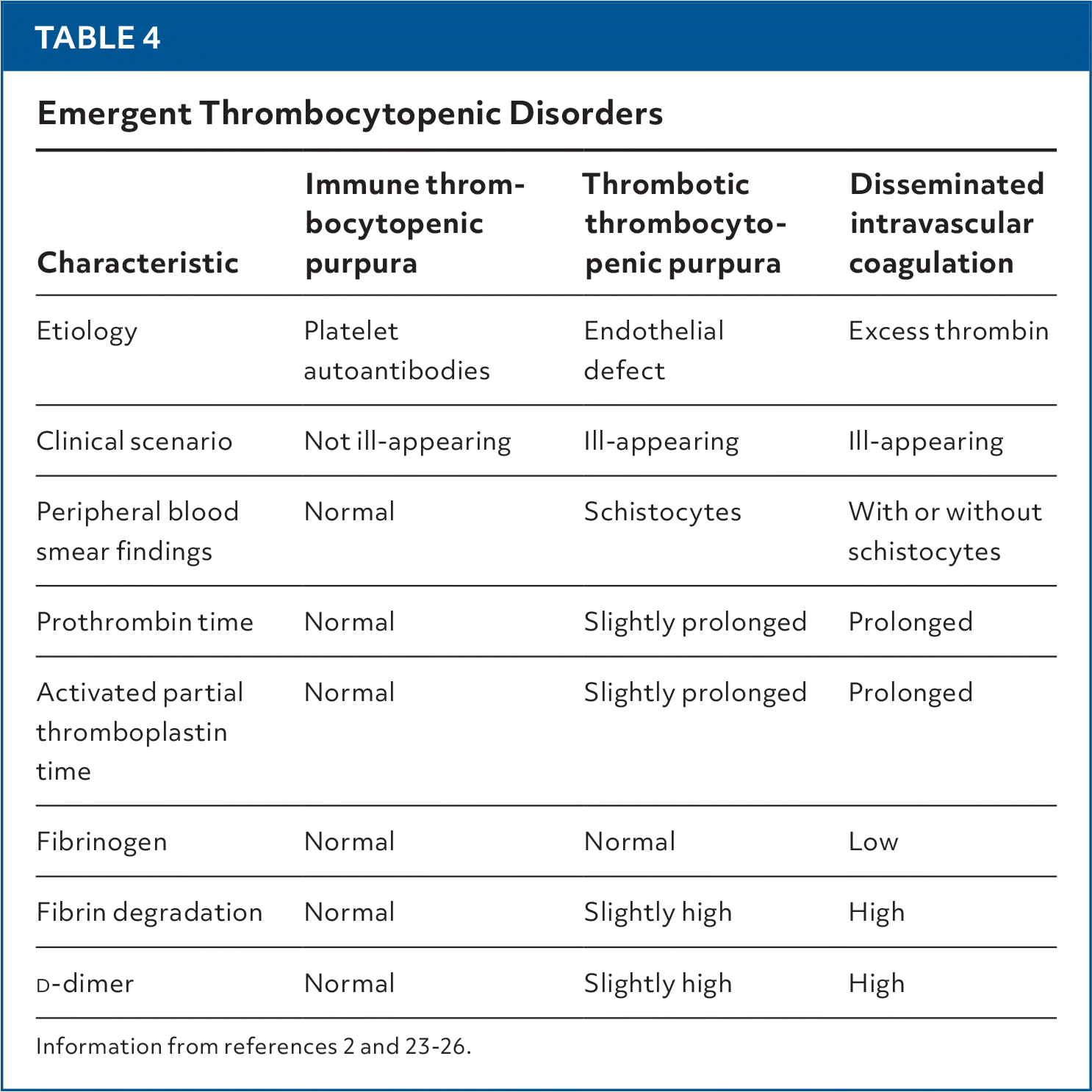
| Characteristic | Immune thrombocytopenic purpura | Thrombotic thrombocytopenic purpura | Disseminated intravascular coagulation |
|---|---|---|---|
| Etiology | Platelet autoantibodies | Endothelial defect | Excess thrombin |
| Clinical scenario | Not ill-appearing | Ill-appearing | Ill-appearing |
| Peripheral blood smear findings | Normal | Schistocytes | With or without schistocytes |
| Prothrombin time | Normal | Slightly prolonged | Prolonged |
| Activated partial thromboplastin time | Normal | Slightly prolonged | Prolonged |
| Fibrinogen | Normal | Normal | Low |
| Fibrin degradation | Normal | Slightly high | High |
| Normal | Slightly high | High |
The American Society of Hematology recommends corticosteroids for most newly diagnosed patients with a platelet count of less than 30 × 103 per μL.23 Corticosteroids and hospitalization are recommended for newly diagnosed patients with a platelet count less than 20 × 103 per μL.27 Administering an IgG infusion immediately after platelet transfusion increases platelet survival.23 See the 2022 American Family Physician article for more on thrombocytopenia.2
Thrombotic Thrombocytopenic Purpura
Thrombotic thrombocytopenic purpura is a rare thrombotic microangiopathy that results in platelet aggregation and microthrombi formation, causing ischemic end-organ injury.28,29 Patients may present with a clinical pentad of thrombocytopenia, hemolytic anemia, acute kidney injury, fever, and neurologic symptoms such as mental status changes, seizures, ataxia, and aphasia. ADAMTS-13 (enzyme that processes von Willebrand factor) deficiency from hereditary mutations or acquired autoantibodies causes thrombotic thrombocytopenic purpura.29
Initial treatment for thrombotic thrombocytopenic purpura includes immunosuppression with corticosteroids and total plasma exchange.30,31 Rituximab and caplacizumab (Cablivi) are adjunctive therapies.24,31,32
Disseminated Intravascular Coagulation
Disseminated intravascular coagulation results from the uninhibited activation of the coagulation cascade by numerous pathways, potentially leading to the development of fibrin deposition, microthrombi formation, bleeding from uncontrolled platelet and clotting factor consumption, and, finally, multiorgan failure.26 Patients with disseminated intravascular coagulation may develop spontaneous gingival bleeding, petechiae, ecchymosis, gastrointestinal bleeding, and oozing from inserted tubes, lines, and drains.
Evaluation of disseminated intravascular coagulation should include obtaining fibrinogen, platelets, d-dimer, PT, and activated PTT and assessing risk using a calculator such as the one created by the International Society on Thrombosis and Haemostasis.33 Treatment for disseminated intravascular coagulation involves addressing the underlying cause and treating potential etiologies such as sepsis, contaminated devices, infected organs, abscesses, or malignancies (Table 525,26,34,35). Patients with disseminated intravascular coagulation and a platelet count of less than 50 × 103 per μL who are bleeding or less than 20 × 103 per μL who are not bleeding should receive platelet transfusions.25,35
TABLE 5. Etiologies of Disseminated Intravascular Coagulation
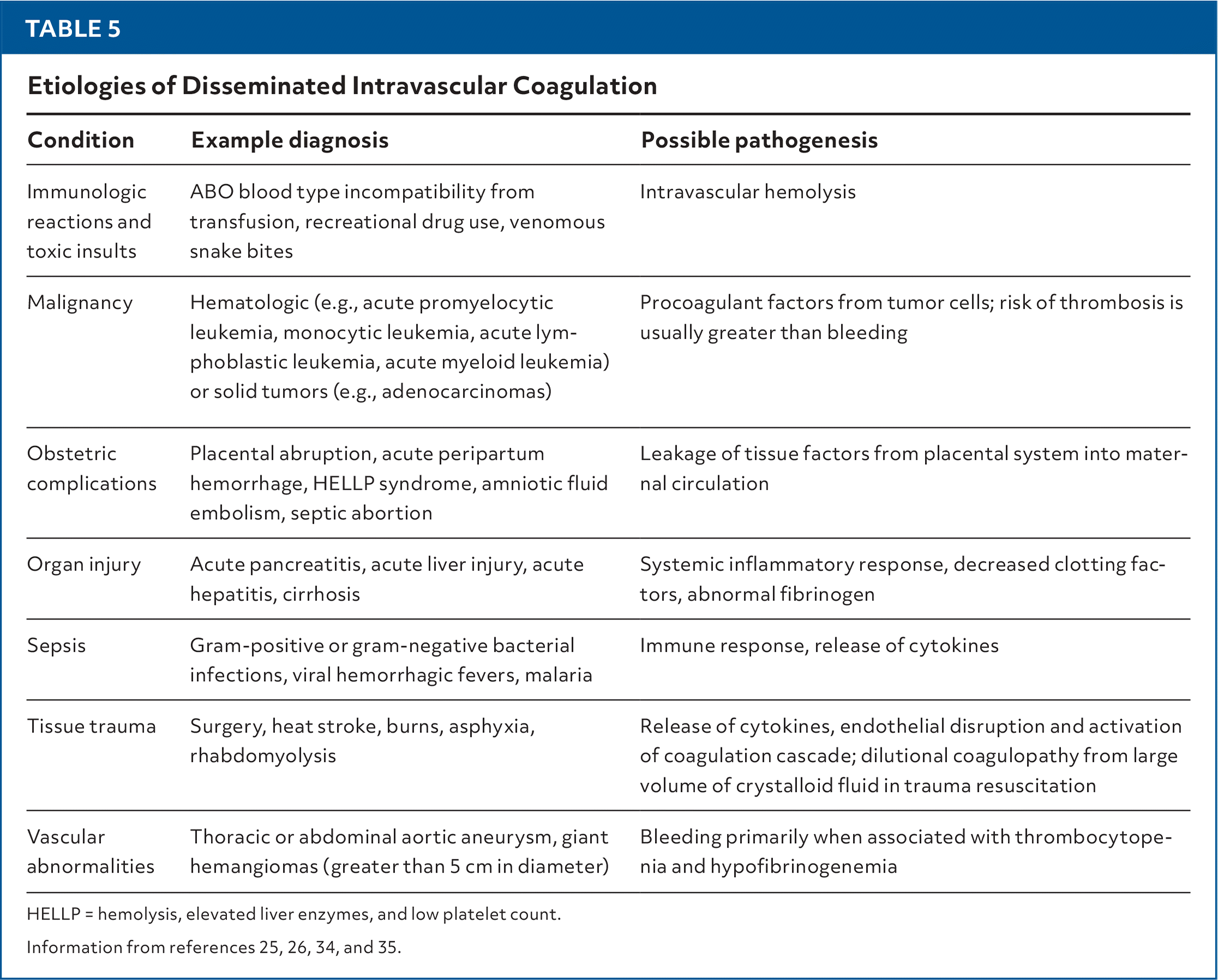
| Condition | Example diagnosis | Possible pathogenesis |
|---|---|---|
| Immunologic reactions and toxic insults | ABO blood type incompatibility from transfusion, recreational drug use, venomous snake bites | Intravascular hemolysis |
| Malignancy | Hematologic (e.g., acute promyelocytic leukemia, monocytic leukemia, acute lymphoblastic leukemia, acute myeloid leukemia) or solid tumors (e.g., adenocarcinomas) | Procoagulant factors from tumor cells; risk of thrombosis is usually greater than bleeding |
| Obstetric complications | Placental abruption, acute peripartum hemorrhage, HELLP syndrome, amniotic fluid embolism, septic abortion | Leakage of tissue factors from placental system into maternal circulation |
| Organ injury | Acute pancreatitis, acute liver injury, acute hepatitis, cirrhosis | Systemic inflammatory response, decreased clotting factors, abnormal fibrinogen |
| Sepsis | Gram-positive or gram-negative bacterial infections, viral hemorrhagic fevers, malaria | Immune response, release of cytokines |
| Tissue trauma | Surgery, heat stroke, burns, asphyxia, rhabdomyolysis | Release of cytokines, endothelial disruption and activation of coagulation cascade; dilutional coagulopathy from large volume of crystalloid fluid in trauma resuscitation |
| Vascular abnormalities | Thoracic or abdominal aortic aneurysm, giant hemangiomas (greater than 5 cm in diameter) | Bleeding primarily when associated with thrombocytopenia and hypofibrinogenemia |
HELLP = hemolysis, elevated liver enzymes, and low platelet count.
Often confused with disseminated intravascular coagulation, trauma coagulopathy, and dilutional coagulopathy resulting from massive transfusion of greater than 10 units of packed RBCs can also cause severe bleeding and are emergencies.36 Trauma coagulopathy should be treated with antifibrinolytic agents, which are contraindicated in disseminated intravascular coagulation.36 Dilutional coagulopathy is treated with cryoprecipitate, platelets, and fresh frozen plasma.
Data Sources: A search was performed in ClinicalKey, Dynamed, Essential Evidence Plus, and PubMed using the key words hematologic emergency, disseminated intravascular coagulation, hemolytic anemia, hemophilia A, hemophilia B, thrombocytopenia, and thrombotic thrombocytopenic purpura. The search included meta-analyses, randomized controlled trials, clinical practice guidelines, and review articles. Search dates: December 2022 to February 2023, and April 2024.
The authors thank Manh Dang, MD, for his review of the manuscript. At the time of submission, Dr. Dang was a hematologist/oncologist at the East Hawaii Cancer Center in Hilo.
The views expressed herein are those of the authors and do not reflect the official policy of the U.S. Department of the Army, U.S. Department of Defense, or U.S. government.
